 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6xsv: X-ray structure of a tetragonal crystal form of alpha amylase fro... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xsv | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of a tetragonal crystal form of alpha amylase from Aspergillus oryzae (Tala-Amylase) at 1.65 A resolution | ||||||
 Components Components | Alpha-amylase | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / substrate complex / Novo lipase / catalytic site / ligands | ||||||
| Function / homology |  Function and homology information Function and homology informationcell wall-bounded periplasmic space / hyphal septin band / spitzenkorper / fungal-type cell wall / alpha-amylase / cell septum / carbohydrate catabolic process / alpha-amylase activity / calcium ion binding / extracellular regionSimilarity search - Function | ||||||
| Biological species |  Aspergillus oryzae (mold) Aspergillus oryzae (mold) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | McPherson, A. | ||||||
 Citation Citation | Journal: J.Biosci.Bioeng. / Year: 2021 Title: Structures of two novel crystal forms of Aspergillus oryzae alpha amylase (taka-amylase). Authors: Gee, C.L. / Holton, J.M. / McPherson, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xsv.cif.gz | 202.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xsv.ent.gz | 159 KB | Display | PDB format |
| PDBx/mmJSON format | 6xsv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xs/6xsvftp://data.pdbj.org/pub/pdb/validation_reports/xs/6xsv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6xsjC  2taaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 54812.746 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Aspergillus oryzae (mold) / Gene: rhaG, amy3, OAory_01056410 / Production host: Aspergillus oryzae (mold)References: UniProt: B0FZ76, UniProt: P0C1B3*PLUS, alpha-amylase |
|---|
-Sugars , 2 types, 4 molecules 
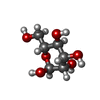
| #2: Sugar | N-Acetylglucosamine Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #4: Sugar | Mannose Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 2 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
|---|
-Non-polymers , 4 types, 517 molecules 



| #3: Chemical | Palmitic acid Mass: 256.424 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H32O2 Mass: 256.424 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H32O2#5: Chemical | ChemComp-MES / | MES (buffer) Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#6: Chemical | ChemComp-CA / |  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 513 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.47 Å3/Da / Density % sol: 50.1 % / Description: elongated rectangular prisms |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: Crystals grown by sitting drop vapor diffusion in Cryschem plates using 0.6 ml reservoirs of 20% PEG 3350 in 0.1 M MES buffer at pH 6.5. Drops composed of equal amounts of the protein at 30 ...Details: Crystals grown by sitting drop vapor diffusion in Cryschem plates using 0.6 ml reservoirs of 20% PEG 3350 in 0.1 M MES buffer at pH 6.5. Drops composed of equal amounts of the protein at 30 mg/ml in water with the reservoir solution. Crystals grew after about 0ne to two weeks at 298 K PH range: 6 - 7.247 |
-Data collection
| Diffraction | Mean temperature: 173 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 1 Å / Beamline: 8.3.1 / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 300K / Detector: PIXEL / Date: Feb 22, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→66.45 Å / Num. obs: 66065 / % possible obs: 99.8 % / Redundancy: 124.3 % / Biso Wilson estimate: 22 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.135 / Rpim(I) all: 0.027 / Rrim(I) all: 0.141 / Rsym value: 0.128 / Net I/σ(I): 16.7 |
| Reflection shell | Resolution: 1.65→1.68 Å / Redundancy: 41 % / Mean I/σ(I) obs: 1.1 / Num. unique obs: 3098 / CC1/2: 0.19 / % possible all: 96.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2TAA Resolution: 1.65→66.45 Å / SU ML: 0.2 / Cross valid method: THROUGHOUT / σ(F): 1.32 / Phase error: 23.22 / Stereochemistry target values: ML Details: Structure was refined to convergence in REFMAC then passed through ten runs of COOT rebuilding and REFINE from PHENIX.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 138.31 Å2 / Biso mean: 43.223 Å2 / Biso min: 20 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.65→66.45 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 24
|
