| Entry | Database: PDB / ID: 6pcc
|
|---|
| Title | Crystal structure of beta-ketoadipyl-CoA thiolase mutant (H356A) in complex hexanoyl coenzyme A |
|---|
 Components Components | Beta-ketoadipyl-CoA thiolase |
|---|
 Keywords Keywords |  TRANSFERASE / Thiolase / aromatic pollutant catabolism / degradative enzymes TRANSFERASE / Thiolase / aromatic pollutant catabolism / degradative enzymes |
|---|
| Function / homology |  Function and homology information Function and homology information
3-oxoadipyl-CoA thiolase / 3-oxoadipyl-CoA thiolase activity / acetyl-CoA C-acyltransferase activity / 3,4-dihydroxybenzoate catabolic process / phenylacetate catabolic process / fatty acid beta-oxidationSimilarity search - Function Beta-ketoadipyl CoA thiolase / Thiolase, active site / Thiolases active site. / Thiolase, acyl-enzyme intermediate active site / Thiolases acyl-enzyme intermediate signature. / Thiolase, conserved site / Thiolases signature 2. / Thiolase / Thiolase, C-terminal / Thiolase, C-terminal domain ...Beta-ketoadipyl CoA thiolase / Thiolase, active site / Thiolases active site. / Thiolase, acyl-enzyme intermediate active site / Thiolases acyl-enzyme intermediate signature. / Thiolase, conserved site / Thiolases signature 2. / Thiolase / Thiolase, C-terminal / Thiolase, C-terminal domain / Thiolase, N-terminal / Thiolase, N-terminal domain / Thiolase-likeSimilarity search - Domain/homology |
|---|
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) |
|---|
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.96 Å |
|---|
 Authors Authors | Sukritee, B. / Panjikar, S. |
|---|
 Citation Citation | Journal: J Struct Biol X / Year: 2020
Title: Structural basis for differentiation between two classes of thiolase: Degradative vs biosynthetic thiolase.
Authors: Bhaskar, S. / Steer, D.L. / Anand, R. / Panjikar, S. |
|---|
| History | | Deposition | Jun 17, 2019 | Deposition site: RCSB / Processing site: RCSB |
|---|
| Revision 1.0 | May 27, 2020 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 22, 2020 | Group: Database references / Category: citation / citation_author
Item: _citation.journal_volume / _citation.page_first ..._citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_PubMed / _citation.title / _citation_author.identifier_ORCID / _citation_author.name |
|---|
| Revision 1.2 | Apr 3, 2024 | Group: Data collection / Database references / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj





 Assembly
Assembly
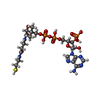
 Mass: 767.534 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H36N7O16P3S / Feature type: SUBJECT OF INVESTIGATION
Mass: 767.534 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H36N7O16P3S / Feature type: SUBJECT OF INVESTIGATION
 Mass: 100.159 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H12O / Feature type: SUBJECT OF INVESTIGATION
Mass: 100.159 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H12O / Feature type: SUBJECT OF INVESTIGATION
 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 1215 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1215 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: MX2 / Wavelength: 0.95 Å
/ Beamline: MX2 / Wavelength: 0.95 Å Processing
Processing