 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6p12: Structure of spastin AAA domain (wild-type) in complex with diami... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6p12 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of spastin AAA domain (wild-type) in complex with diaminotriazole-based inhibitor | |||||||||
 Components Components | Drosophila melanogaster Spastin AAA domain | |||||||||
 Keywords Keywords | ISOMERASE/ISOMERASE INHIBITOR /  inhibitor / complex / AAA protein / ISOMERASE-ISOMERASE INHIBITOR complex inhibitor / complex / AAA protein / ISOMERASE-ISOMERASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of axon extension involved in regeneration / hemocyte migration / negative regulation of synaptic assembly at neuromuscular junction / Sealing of the nuclear envelope (NE) by ESCRT-III / negative regulation of neuromuscular synaptic transmission / positive regulation of neuromuscular synaptic transmission / microtubule-severing ATPase / positive regulation of synaptic assembly at neuromuscular junction / microtubule severing ATPase activity / mitotic chromosome movement towards spindle pole ...positive regulation of axon extension involved in regeneration / hemocyte migration / negative regulation of synaptic assembly at neuromuscular junction / Sealing of the nuclear envelope (NE) by ESCRT-III / negative regulation of neuromuscular synaptic transmission / positive regulation of neuromuscular synaptic transmission / microtubule-severing ATPase / positive regulation of synaptic assembly at neuromuscular junction / microtubule severing ATPase activity / mitotic chromosome movement towards spindle pole / microtubule severing / regulation of terminal button organization / positive regulation of lipid metabolic process / positive regulation of microtubule depolymerization / mitotic spindle elongation / negative regulation of microtubule depolymerization / protein hexamerization / positive regulation of dendrite morphogenesis / mitotic sister chromatid segregation / alpha-tubulin binding / isomerase activity / lipid droplet / adult locomotory behavior / locomotory behavior / neuromuscular junction / terminal bouton / microtubule cytoskeleton organization / spindle / microtubule cytoskeleton / chromosome / nervous system development / microtubule binding / microtubule / cell division / centrosome / ATP hydrolysis activity / ATP binding / membrane / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Drosophila melanogaster (fruit fly) Drosophila melanogaster (fruit fly) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9413185678 Å | |||||||||
 Authors Authors | Pisa, R. / Cupido, T. / Kapoor, T.M. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Cell Chem Biol / Year: 2019 Title: Analyzing Resistance to Design Selective Chemical Inhibitors for AAA Proteins. Authors: Pisa, R. / Cupido, T. / Steinman, J.B. / Jones, N.H. / Kapoor, T.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6p12.cif.gz | 92 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6p12.ent.gz | 53.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6p12.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p1/6p12ftp://data.pdbj.org/pub/pdb/validation_reports/p1/6p12 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6p10C  6p11C  6p13C  6p14C  3b9pS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34191.848 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Drosophila melanogaster (fruit fly) / Gene: spas, CG5977 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q8I0P1, microtubule-severing ATPase Escherichia coli (E. coli) / References: UniProt: Q8I0P1, microtubule-severing ATPase |
|---|---|
| #2: Chemical | ChemComp-NKY / 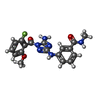  Mass: 384.364 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H17FN6O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 384.364 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H17FN6O3 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Chemical | ChemComp-MPD / (2-Methyl-2,4-pentanediol  Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 218 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 218 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.65 Å3/Da / Density % sol: 57.88 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop Details: 0.1 M sodium acetate, pH 5.0-7.0, 2% PEG4000, 15% MPD PH range: 5.0-7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS-II / Beamline: 17-ID-1 / Wavelength: 0.9197 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Jul 14, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9197 Å / Relative weight: 1 |
| Reflection | Resolution: 1.94→50 Å / Num. obs: 26363 / % possible obs: 99 % / Redundancy: 6.5 % / Biso Wilson estimate: 43.2284817795 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.031 / Net I/σ(I): 16.6 |
| Reflection shell | Resolution: 1.94→1.97 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 2.8 / Num. unique obs: 1323 / CC1/2: 0.932 / % possible all: 99.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3B9P Resolution: 1.9413185678→40.336 Å / SU ML: 0.256993797688 / Cross valid method: FREE R-VALUE / σ(F): 1.38937703476 / Phase error: 25.154924059
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.7947301096 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9413185678→40.336 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
