 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
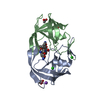
Yorodumi- PDB-6oyr: X-ray crystal structure of wild type HIV-1 protease in complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6oyr | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray crystal structure of wild type HIV-1 protease in complex with GRL-002 | ||||||
 Components Components | Protease | ||||||
 Keywords Keywords | VIRAL PROTEIN/INHIBITOR / Inhibitor / Viral protein / Viral protein-Inhibitor Complex | ||||||
| Function / homology |  Function and homology information: / : / HIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA ...: / : / HIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA / viral penetration into host nucleus / establishment of integrated proviral latency / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / symbiont-mediated suppression of host gene expression / viral nucleocapsid / DNA recombination / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / DNA-directed DNA polymerase activity / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / RNA binding / zinc ion binding / membrane Function and homology information: / : / HIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA ...: / : / HIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA / viral penetration into host nucleus / establishment of integrated proviral latency / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / symbiont-mediated suppression of host gene expression / viral nucleocapsid / DNA recombination / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / DNA-directed DNA polymerase activity / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / RNA binding / zinc ion binding / membraneSimilarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.54 Å | ||||||
 Authors Authors | Bulut, H. / Hattori, S.I. / Aoki-Ogata, H. / Hayashi, H. / Aoki, M. / Ghosh, A.K. / Mitsuya, H. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Sci Rep / Year: 2020 Title: Single atom changes in newly synthesized HIV protease inhibitors reveal structural basis for extreme affinity, high genetic barrier, and adaptation to the HIV protease plasticity. Authors: Bulut, H. / Hattori, S.I. / Aoki-Ogata, H. / Hayashi, H. / Das, D. / Aoki, M. / Davis, D.A. / Rao, K.V. / Nyalapatla, P.R. / Ghosh, A.K. / Mitsuya, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6oyr.cif.gz | 37.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6oyr.ent.gz | 23.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6oyr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oy/6oyrftp://data.pdbj.org/pub/pdb/validation_reports/oy/6oyr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6oglC  6ogpC  6ogqC  6ogsC  6ogtC  6oydC  5tyrS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 10830.781 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Gene: pol / Production host:  Escherichia coli (E. coli) / References: UniProt: Q8ULI2, UniProt: P12497*PLUS Escherichia coli (E. coli) / References: UniProt: Q8ULI2, UniProt: P12497*PLUS |
|---|---|
| #2: Chemical | ChemComp-NJY / (  Mass: 674.780 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H43FN4O8S / Feature type: SUBJECT OF INVESTIGATION Mass: 674.780 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H43FN4O8S / Feature type: SUBJECT OF INVESTIGATION |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 43 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.2 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: 0.15 M Ammonium sulfate, 0.1 M HEPES (pH 7.4), 15% (w/v) PEG4000 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL41XU / Wavelength: 1 Å / Beamline: BL41XU / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 31, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.54→45.39 Å / Num. obs: 14722 / % possible obs: 99.32 % / Redundancy: 18.6 % / Net I/σ(I): 15.72 |
| Reflection shell | Resolution: 1.54→1.595 Å / Num. unique obs: 14710 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5TYR Resolution: 1.54→45.39 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.944 / SU B: 2.374 / SU ML: 0.082 / Cross valid method: THROUGHOUT / ESU R: 0.097 / ESU R Free: 0.102 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.835 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.54→45.39 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|