 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ftp: Crystal form 1 of Alpha1-antichymotrypsin variant DBS-II-allo: an... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ftp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal form 1 of Alpha1-antichymotrypsin variant DBS-II-allo: an allosterically modulated drug-binding serpin for doxorubicin | ||||||
 Components Components | (Alpha-1-antichymotrypsin Alpha 1-antichymotrypsin) x 2 Alpha 1-antichymotrypsin) x 2 | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Serpin / alpha1-Antichymotrypsin / Doxorubicin-binding protein / allosterically triggered drug release | ||||||
| Function / homology |  Function and homology information Function and homology informationmaintenance of gastrointestinal epithelium / regulation of lipid metabolic process / platelet alpha granule lumen / acute-phase response / serine-type endopeptidase inhibitor activity / azurophil granule lumen / Platelet degranulation / collagen-containing extracellular matrix / secretory granule lumen / blood microparticle ...maintenance of gastrointestinal epithelium / regulation of lipid metabolic process / platelet alpha granule lumen / acute-phase response / serine-type endopeptidase inhibitor activity / azurophil granule lumen / Platelet degranulation / collagen-containing extracellular matrix / secretory granule lumen / blood microparticle / inflammatory response / Neutrophil degranulation / DNA binding / extracellular space / extracellular exosome / extracellular region / nucleusSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Schmidt, K. / Muller, Y.A. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: Proc. Natl. Acad. Sci. U.S.A. / Year: 2018 Title: Design of an allosterically modulated doxycycline and doxorubicin drug-binding protein. Authors: Schmidt, K. / Gardill, B.R. / Kern, A. / Kirchweger, P. / Borsch, M. / Muller, Y.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ftp.cif.gz | 100.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ftp.ent.gz | 74.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6ftp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ft/6ftpftp://data.pdbj.org/pub/pdb/validation_reports/ft/6ftp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5om2C  5om3C  5om5C  5om6C  5om7C  5om8SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Alpha 1-antichymotrypsin / ACT / Cell growth-inhibiting gene 24/25 protein / Serpin A3 Mass: 41986.516 Da / Num. of mol.: 1 Mutation: L24R W194F W215Y E242Q K244N L269S P270Q K274S W276F R277F D278E A349R V355L K356E I357V T358L L359F L360Q Source method: isolated from a genetically manipulated source Details: All N-terminal residues that are present in the sample sequence but not in the PDB file could not be modelled due to missing electron density. The protein is splitted into chain A and chain ...Details: All N-terminal residues that are present in the sample sequence but not in the PDB file could not be modelled due to missing electron density. The protein is splitted into chain A and chain B, as it belongs to the serine proteinase inhibitor (serpin) family and is proteolytically cleaved C-terminal of Gln360. The residues Gln360 and Arg376, which is the first residue visible in the electron density, are 70 angstrom apart because of the serpin-typical conformational change upon proteolytical cleavage. The sequence following the P1-P1' scissile bond, namely residues GPL..KQA, are part of chain B. Source: (gene. exp.) Homo sapiens (human) / Gene: SERPINA3, AACT, GIG24, GIG25 / Production host:  Escherichia coli BL21(DE3) (bacteria) / Variant (production host): Star / References: UniProt: P01011 Escherichia coli BL21(DE3) (bacteria) / Variant (production host): Star / References: UniProt: P01011 | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | Alpha 1-antichymotrypsin / ACT / Cell growth-inhibiting gene 24/25 protein / Serpin A3 Mass: 4720.580 Da / Num. of mol.: 1 / Mutation: S361G A362P P382D T383N D384F Q386W N387S Source method: isolated from a genetically manipulated source Details: The protein is splitted into chain A and chain B, as it belongs to the serine proteinase inhibitor (serpin) family and is proteolytically cleaved C-terminal of Gln360. The residues Gln360 ...Details: The protein is splitted into chain A and chain B, as it belongs to the serine proteinase inhibitor (serpin) family and is proteolytically cleaved C-terminal of Gln360. The residues Gln360 and Arg376, which is the first residue visible in the electron density, are 70 angstrom apart because of the serpin-typical conformational change upon proteolytical cleavage. The sequence following the P1-P1' scissile bond, namely residues GPL..KQA, are part of chain B. The residues 361-366 (GPLVET) are missing in the PDB file because of missing electron density. Source: (gene. exp.) Homo sapiens (human) / Gene: SERPINA3, AACT, GIG24, GIG25 / Production host: Escherichia coli BL21(DE3) (bacteria) / Variant (production host): Star / References: UniProt: P01011 | ||||
| #3: Chemical | Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-DM2 / | Doxorubicin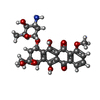  Mass: 543.519 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H29NO11 / Comment: medication, chemotherapy*YM Mass: 543.519 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H29NO11 / Comment: medication, chemotherapy*YM#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.44 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / Details: 0.2 M sodium thiocyanate, 20 % w/v PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY / Beamline: 14.2 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: May 12, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→42.12 Å / Num. obs: 37572 / % possible obs: 99.9 % / Redundancy: 9.9 % / CC1/2: 1 / Rrim(I) all: 0.061 / Rsym value: 0.058 / Net I/σ(I): 25.31 |
| Reflection shell | Resolution: 1.8→1.91 Å / Redundancy: 10.1 % / Mean I/σ(I) obs: 2.76 / Num. unique obs: 5991 / CC1/2: 0.854 / Rrim(I) all: 0.878 / Rsym value: 0.834 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5OM8 Resolution: 1.8→42.12 Å / SU ML: 0.22 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 23.34
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→42.12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
