 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6f0g | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure ASF1-ip3 | |||||||||||||||
 Components Components |
| |||||||||||||||
 Keywords Keywords |  CHAPERONE / protein-peptide complexe CHAPERONE / protein-peptide complexe | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationhistone chaperone activity / muscle cell differentiation / DNA replication-dependent chromatin assembly / DNA repair-dependent chromatin remodeling / Formation of Senescence-Associated Heterochromatin Foci (SAHF) / replication fork processing / osteoblast differentiation / nucleosome assembly / site of double-strand break / histone binding ...histone chaperone activity / muscle cell differentiation / DNA replication-dependent chromatin assembly / DNA repair-dependent chromatin remodeling / Formation of Senescence-Associated Heterochromatin Foci (SAHF) / replication fork processing / osteoblast differentiation / nucleosome assembly / site of double-strand break / histone binding / DNA repair / chromatin binding / chromatin / protein-containing complex / nucleoplasm / nucleusSimilarity search - Function | |||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.3 Å | |||||||||||||||
 Authors Authors | Gaubert, A. / Guichard, B. / Richet, N. / Le Du, M.H. / Andreani, J. / Guerois, R. / Ochsenbein, F. | |||||||||||||||
| Funding support |  France, 4items France, 4items
| |||||||||||||||
 Citation Citation | Journal: Cell Chem Biol / Year: 2019 Title: Design on a Rational Basis of High-Affinity Peptides Inhibiting the Histone Chaperone ASF1. Authors: Bakail, M. / Gaubert, A. / Andreani, J. / Moal, G. / Pinna, G. / Boyarchuk, E. / Gaillard, M.C. / Courbeyrette, R. / Mann, C. / Thuret, J.Y. / Guichard, B. / Murciano, B. / Richet, N. / ...Authors: Bakail, M. / Gaubert, A. / Andreani, J. / Moal, G. / Pinna, G. / Boyarchuk, E. / Gaillard, M.C. / Courbeyrette, R. / Mann, C. / Thuret, J.Y. / Guichard, B. / Murciano, B. / Richet, N. / Poitou, A. / Frederic, C. / Le Du, M.H. / Agez, M. / Roelants, C. / Gurard-Levin, Z.A. / Almouzni, G. / Cherradi, N. / Guerois, R. / Ochsenbein, F. | |||||||||||||||
| History |
|
- Structure visualization
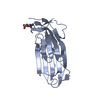
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6f0g.cif.gz | 158.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6f0g.ent.gz | 126.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6f0g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f0/6f0gftp://data.pdbj.org/pub/pdb/validation_reports/f0/6f0g | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 17927.998 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ASF1A, CGI-98, HSPC146 / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 DE3 / References: UniProt: Q9Y294 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 DE3 / References: UniProt: Q9Y294#2: Protein/peptide | Mass: 2779.144 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: rational design / Source: (synth.) Homo sapiens (human)#3: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.3 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / Details: Citrate 0.1M pH 2.5, LiSO4 0.25M, PEG8000 7% |
-Data collection
| Diffraction | Mean temperature: 80 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL / Beamline: PROXIMA 1 / Wavelength: 0.98011 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Mar 8, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98011 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→45 Å / Num. obs: 16434 / % possible obs: 99.6 % / Redundancy: 7.9 % / Biso Wilson estimate: 28.48 Å2 / Rmerge(I) obs: 0.025 / Net I/σ(I): 8.3 |
| Reflection shell | Resolution: 2.3→2.44 Å / Redundancy: 7.6 % / Rmerge(I) obs: 0.1299 / % possible all: 97.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.3→38.88 Å / Cor.coef. Fo:Fc: 0.9074 / Cor.coef. Fo:Fc free: 0.8756 / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.03 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.303 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.3→38.88 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.33 Å / Total num. of bins used: 9
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
