Entry Database : PDB / ID : 6es8Title HIV capsid hexamer with IP6 ligand Gag protein Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Method / / / Resolution : 1.9 Å Authors James, L.C. Journal : Elife / Year : 2018Title : IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis.Authors : Mallery, D.L. / Marquez, C.L. / McEwan, W.A. / Dickson, C.F. / Jacques, D.A. / Anandapadamanaban, M. / Bichel, K. / Towers, G.J. / Saiardi, A. / Bocking, T. / James, L.C. History Deposition Oct 19, 2017 Deposition site / Processing site Revision 1.0 Aug 15, 2018 Provider / Type Revision 1.1 Oct 7, 2020 Group / Structure summary / Category / struct_connItem _chem_comp.pdbx_synonyms / _struct_conn.pdbx_dist_value ... _chem_comp.pdbx_synonyms / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_ptnr1_label_alt_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry Revision 1.2 Jan 17, 2024 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components HIV-1 protease
HIV-1 protease  Keywords
Keywords Function and homology information
Function and homology information

 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj




 Assembly
Assembly


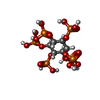
 Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6
Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 Mass: 18.015 Da / Num. of mol.: 120 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 120 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I02 / Wavelength: 1 Å
/ Beamline: I02 / Wavelength: 1 Å Processing
Processing