 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6dim: Crystal structure of Tdp1 catalytic domain in complex with Zenobi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6dim | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Tdp1 catalytic domain in complex with Zenobia fragment ZT1982 from cocktail soak | ||||||
 Components Components | Tyrosyl-DNA phosphodiesterase 1 | ||||||
 Keywords Keywords | hydrolase/hydrolase inhibitor / fragment-based drug design / hydrolase-hydrolase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology information3'-tyrosyl-DNA phosphodiesterase activity / single strand break repair /  Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / exonuclease activity / Nonhomologous End-Joining (NHEJ) / double-strand break repair / single-stranded DNA binding / double-stranded DNA binding / intracellular membrane-bounded organelle / DNA repair ...3'-tyrosyl-DNA phosphodiesterase activity / single strand break repair / Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / exonuclease activity / Nonhomologous End-Joining (NHEJ) / double-strand break repair / single-stranded DNA binding / double-stranded DNA binding / intracellular membrane-bounded organelle / DNA repair / nucleoplasm / nucleus / plasma membrane / cytoplasm Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / exonuclease activity / Nonhomologous End-Joining (NHEJ) / double-strand break repair / single-stranded DNA binding / double-stranded DNA binding / intracellular membrane-bounded organelle / DNA repair ...3'-tyrosyl-DNA phosphodiesterase activity / single strand break repair / Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases / exonuclease activity / Nonhomologous End-Joining (NHEJ) / double-strand break repair / single-stranded DNA binding / double-stranded DNA binding / intracellular membrane-bounded organelle / DNA repair / nucleoplasm / nucleus / plasma membrane / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.81 Å | ||||||
 Authors Authors | Lountos, G.T. / Zhao, X.Z. / Kiselev, E. / Tropea, J.E. / Needle, D. / Burke Jr., T.R. / Pommier, Y. / Waugh, D.S. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2019 Title: Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening. Authors: Lountos, G.T. / Zhao, X.Z. / Kiselev, E. / Tropea, J.E. / Needle, D. / Pommier, Y. / Burke, T.R. / Waugh, D.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6dim.cif.gz | 206.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6dim.ent.gz | 162.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6dim.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/di/6dimftp://data.pdbj.org/pub/pdb/validation_reports/di/6dim | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6dhuC  6dieC  6dihC  6djdC  6djeC  6djfC  6djgC  6djhC  6djiC  6djjC  6mj5C  6n17C  6n19C C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 52126.336 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TDP1 / Production host:  Escherichia coli (E. coli) Escherichia coli (E. coli)References: UniProt: Q9NUW8, Hydrolases; Acting on ester bonds; Phosphoric-diester hydrolases#2: Chemical | 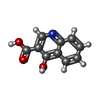  Mass: 189.167 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H7NO3 Mass: 189.167 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H7NO3#3: Chemical | ChemComp-EDO / Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 582 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 582 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.47 Å3/Da / Density % sol: 50.3 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 0.1 M MOPS/HEPES-Na pH 7.5 10% w/v PEG 8000 20% v/v ethylene glycol 0.03M sodium fluoride 0.03M sodium bromide 0.03M sodium iodide |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-BM / Wavelength: 1 Å / Beamline: 22-BM / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Mar 13, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.81→50 Å / Num. obs: 93819 / % possible obs: 97.8 % / Redundancy: 4.2 % / Rmerge(I) obs: 0.071 / Rpim(I) all: 0.036 / Net I/σ(I): 29.2 |
| Reflection shell | Resolution: 1.82→1.84 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.882 / Mean I/σ(I) obs: 2.3 / Num. unique obs: 4647 / CC1/2: 0.622 / Rpim(I) all: 0.501 / % possible all: 98.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.81→40.988 Å / SU ML: 0.21 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 24.08
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.81→40.988 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
