 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6b8w: 1.9 Angstrom Resolution Crystal Structure of Cupin_2 Domain (pfam... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6b8w | ||||||
|---|---|---|---|---|---|---|---|
| Title | 1.9 Angstrom Resolution Crystal Structure of Cupin_2 Domain (pfam 07883) of XRE Family Transcriptional Regulator from Enterobacter cloacae. | ||||||
 Components Components | XRE family transcriptional regulator | ||||||
 Keywords Keywords |  TRANSCRIPTION / Structural Genomics / Center for Structural Genomics of Infectious Diseases / CSGID / XRE Family Transcriptional Regulator. TRANSCRIPTION / Structural Genomics / Center for Structural Genomics of Infectious Diseases / CSGID / XRE Family Transcriptional Regulator. | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Enterobacter cloacae subsp. cloacae (bacteria) Enterobacter cloacae subsp. cloacae (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Minasov, G. / Wawrzak, Z. / Skarina, T. / McChesney, C. / Grimshaw, S. / Sandoval, J. / Satchell, K.J.F. / Savchenko, A. / Joachimiak, A. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
 Citation Citation | Journal: To Be Published Title: 1.9 Angstrom Resolution Crystal Structure of Cupin_2 Domain (pfam 07883) of XRE Family Transcriptional Regulator from Enterobacter cloacae. Authors: Minasov, G. / Wawrzak, Z. / Skarina, T. / McChesney, C. / Grimshaw, S. / Sandoval, J. / Satchell, K.J.F. / Savchenko, A. / Joachimiak, A. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6b8w.cif.gz | 100.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6b8w.ent.gz | 76.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6b8w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b8/6b8wftp://data.pdbj.org/pub/pdb/validation_reports/b8/6b8w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1y9qS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 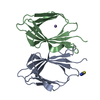
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10807.256 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacter cloacae subsp. cloacae (strain ATCC 13047 / DSM 30054 / NBRC 13535 / NCDC 279-56) (bacteria)Strain: ATCC 13047 / DSM 30054 / NBRC 13535 / NCDC 279-56 / Gene: ECL_02024 / Plasmid: pMCSG53 / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3)-Magic / References: UniProt: A0A0H3CJW2#2: Chemical | ChemComp-SCN / | Thiocyanate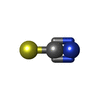  Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS#3: Chemical | ChemComp-MN / |   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 285 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 285 / Source method: isolated from a natural source / Formula: H2OSequence details | Authors state that the full length protein used in study was hydrolyzed during crystallization ...Authors state that the full length protein used in study was hydrolyzed during crystallization experiments, the model starts at 81th residue GLN of UniProt reference, but it is not known exactly where the protein was hydrolyzed. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 58.6 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: Protein: 12.0 mg/ml, 0.3M Sodium chloride, 0.01M HEPES pH 7.5; Screen: 0.2M Sodium thiocyan, 20% (w/v) PEG 3350. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-F / Wavelength: 0.97872 Å / Beamline: 21-ID-F / Wavelength: 0.97872 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Aug 12, 2017 / Details: C(111) |
| Radiation | Monochromator: Be / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97872 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→30 Å / Num. obs: 21115 / % possible obs: 99.5 % / Observed criterion σ(I): -3 / Redundancy: 6.3 % / Biso Wilson estimate: 29.5 Å2 / Rmerge(I) obs: 0.074 / Rpim(I) all: 0.032 / Rrim(I) all: 0.081 / Rsym value: 0.074 / Χ2: 1.028 / Net I/σ(I): 22.1 |
| Reflection shell | Resolution: 1.9→1.93 Å / Redundancy: 5.1 % / Rmerge(I) obs: 0.779 / Mean I/σ(I) obs: 2.2 / Num. unique obs: 1019 / CC1/2: 0.678 / Rpim(I) all: 0.381 / Rrim(I) all: 0.87 / Rsym value: 0.779 / Χ2: 1.029 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1Y9Q Resolution: 1.9→29.87 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.944 / SU B: 6.538 / SU ML: 0.102 / Cross valid method: THROUGHOUT / ESU R: 0.137 / ESU R Free: 0.131 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.6 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.197 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.9→29.87 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|