| Entry | Database: PDB / ID: 6a50
|
|---|
| Title | structure of benzoylformate decarboxylases in complex with cofactor TPP |
|---|
 Components Components | benzoylformate decarboxylases |
|---|
 Keywords Keywords |  LYASE / ThDP / Glycolaldehyde Synthase LYASE / ThDP / Glycolaldehyde Synthase |
|---|
| Function / homology | Thiamin diphosphate (ThDP)-binding fold, Pyr/PP domains / TPP-binding domain / Rossmann fold / 3-Layer(aba) Sandwich / Alpha Beta / THIAMINE DIPHOSPHATE Function and homology information Function and homology information |
|---|
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) |
|---|
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å |
|---|
 Authors Authors | Guo, Y. / Wang, S. / Nie, Y. / Li, S. |
|---|
| Funding support |  China, 2items China, 2items | Organization | Grant number | Country |
|---|
| Chinese Academy of Sciences | ZDRW-ZS-2016 | China | | National Basic Research Program of China (973 Program) | 2015CB755704 | China |
|
|---|
 Citation Citation | Journal: Nat Commun / Year: 2019
Title: A Synthetic Pathway for Acetyl-Coenzyme A Biosynthesis
Authors: Lu, X. / Liu, Y. / Yang, Y. / Wang, S. / Wang, Q. / Wang, X. / Yan, Z. / Cheng, J. / Liu, C. / Yang, X. / Luo, H. / Yang, S. / Gou, J. / Ye, L. / Lu, L. / Zhang, Z. / Guo, Y. / Nie, Y. / ...Authors: Lu, X. / Liu, Y. / Yang, Y. / Wang, S. / Wang, Q. / Wang, X. / Yan, Z. / Cheng, J. / Liu, C. / Yang, X. / Luo, H. / Yang, S. / Gou, J. / Ye, L. / Lu, L. / Zhang, Z. / Guo, Y. / Nie, Y. / Lin, J. / Li, S. / Cai, T. / Ma, H. / Wang, W. / Liu, Y. / Li, Y. / Jiang, H. / Tian, C. |
|---|
| History | | Deposition | Jun 21, 2018 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Feb 20, 2019 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 22, 2023 | Group: Author supporting evidence / Data collection ...Author supporting evidence / Data collection / Database references / Derived calculations / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_audit_support / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_audit_support.funding_organization / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj

 Assembly
Assembly

 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg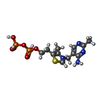
 Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S
Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 18.015 Da / Num. of mol.: 1001 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1001 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing