 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5yhv: Crystal structure of an aminotransferase from Mycobacterium tuber... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5yhv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of an aminotransferase from Mycobacterium tuberculosis | ||||||
 Components Components | Aminotransferase Transaminase Transaminase | ||||||
 Keywords Keywords | TRANSFERASE / Aminotransferase / PLP-dependent / Amino acid metabolism / Cytoplasm / BIOSYNTHETIC PROTEIN | ||||||
| Function / homology |  Function and homology informationvaline-pyruvate transaminase / valine-pyruvate transaminase activity / cell wall / biosynthetic process / pyridoxal phosphate binding Function and homology informationvaline-pyruvate transaminase / valine-pyruvate transaminase activity / cell wall / biosynthetic process / pyridoxal phosphate bindingSimilarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Saroj, D.C. / Biswal, B.K. | ||||||
| Funding support |  India, 1items India, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of an aminotransferase from Mycobacterium tuberculosis Authors: Saroj, D.C. / Biswal, B.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5yhv.cif.gz | 287.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5yhv.ent.gz | 228.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5yhv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yh/5yhvftp://data.pdbj.org/pub/pdb/validation_reports/yh/5yhv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3h14S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 0 / Refine code: 0
NCS ensembles :
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Transaminase Mass: 41940.375 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (bacteria)Strain: ATCC 25618 / H37Rv / Gene: aspB, Rv3565 / Production host: Mycobacterium smegmatis (bacteria)References: UniProt: P96847, Transferases; Transferring nitrogenous groups; Transaminases |
|---|
-Non-polymers , 5 types, 211 molecules 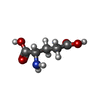



| #2: Chemical | ChemComp-GLU / Glutamic acid Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4 Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4 | ||||||
|---|---|---|---|---|---|---|---|
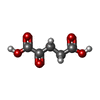
| #3: Chemical | Glycerol Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-PLP / Pyridoxal phosphate Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P#5: Chemical | Α-Ketoglutaric acid Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 201 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.64 % / Description: Rod shaped |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / Details: 0.2M Ammonium phosphate, 0.1M Calcium Chloride |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E+ SUPERBRIGHT / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Sep 5, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→99.39 Å / Num. obs: 50648 / % possible obs: 99.3 % / Redundancy: 4 % / Net I/σ(I): 12.75 |
| Reflection shell | Resolution: 2.7→2.75 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3H14 Resolution: 2.7→99.39 Å / Cor.coef. Fo:Fc: 0.921 / Cor.coef. Fo:Fc free: 0.89 / SU B: 15.673 / SU ML: 0.311 / Cross valid method: THROUGHOUT / ESU R: 1.121 / ESU R Free: 0.36 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.898 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.7→99.39 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|