 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5wp3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of EED in complex with EB22 | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  GENE REGULATION / Structural Genomics / Structural Genomics Consortium / SGC GENE REGULATION / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationESC/E(Z) complex / spinal cord development / histone methyltransferase activity / Transcriptional Regulation by E2F6 / nucleosome binding / enzyme activator activity / transcription corepressor binding / PRC2 methylates histones and DNA / Regulation of PTEN gene transcription / Defective pyroptosis ...ESC/E(Z) complex / spinal cord development / histone methyltransferase activity / Transcriptional Regulation by E2F6 / nucleosome binding / enzyme activator activity / transcription corepressor binding / PRC2 methylates histones and DNA / Regulation of PTEN gene transcription / Defective pyroptosis / PKMTs methylate histone lysines / Activation of anterior HOX genes in hindbrain development during early embryogenesis / HCMV Early Events / chromosome / Oxidative Stress Induced Senescence / negative regulation of DNA-templated transcription / negative regulation of transcription by RNA polymerase II / nucleoplasm / identical protein binding / nucleus / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.55 Å | ||||||
 Authors Authors | Dong, C. / Tempel, W. / Zhu, L. / Moody, J.D. / Baker, D. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Proc. Natl. Acad. Sci. U.S.A. / Year: 2017 Title: First critical repressive H3K27me3 marks in embryonic stem cells identified using designed protein inhibitor. Authors: Moody, J.D. / Levy, S. / Mathieu, J. / Xing, Y. / Kim, W. / Dong, C. / Tempel, W. / Robitaille, A.M. / Dang, L.T. / Ferreccio, A. / Detraux, D. / Sidhu, S. / Zhu, L. / Carter, L. / Xu, C. / ...Authors: Moody, J.D. / Levy, S. / Mathieu, J. / Xing, Y. / Kim, W. / Dong, C. / Tempel, W. / Robitaille, A.M. / Dang, L.T. / Ferreccio, A. / Detraux, D. / Sidhu, S. / Zhu, L. / Carter, L. / Xu, C. / Valensisi, C. / Wang, Y. / Hawkins, R.D. / Min, J. / Moon, R.T. / Orkin, S.H. / Baker, D. / Ruohola-Baker, H. | ||||||
| History |
|
- Structure visualization
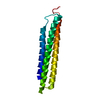
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5wp3.cif.gz | 188.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5wp3.ent.gz | 147 KB | Display | PDB format |
| PDBx/mmJSON format | 5wp3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wp/5wp3ftp://data.pdbj.org/pub/pdb/validation_reports/wp/5wp3 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / hEED / Embryonic ectoderm development protein / WD protein associating with integrin cytoplasmic ...hEED / Embryonic ectoderm development protein / WD protein associating with integrin cytoplasmic tails 1 / WAIT-1 Mass: 42443.324 Da / Num. of mol.: 1 / Fragment: residues 75-401 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: EED / Plasmid: pET28GST-LIC / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3)-V3R / References: UniProt: O75530 Escherichia coli (E. coli) / Strain (production host): BL21(DE3)-V3R / References: UniProt: O75530 |
|---|---|
| #2: Protein | Mass: 13993.254 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Plasmid: pET29b / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3) |
| #3: Chemical | ChemComp-UNX /   Num. of mol.: 16 / Source method: obtained synthetically Num. of mol.: 16 / Source method: obtained synthetically |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41.51 % / Mosaicity: 0.35 ° |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / Details: 20% PEG-3350, 0.2 M sodium bromide |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-E / Wavelength: 0.9792 Å / Beamline: 24-ID-E / Wavelength: 0.9792 Å | ||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Mar 12, 2016 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.5→57.07 Å / Num. obs: 17102 / % possible obs: 99.8 % / Redundancy: 7 % / Biso Wilson estimate: 62.35 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.116 / Rpim(I) all: 0.047 / Rrim(I) all: 0.125 / Net I/σ(I): 14.9 / Num. measured all: 119016 / Scaling rejects: 0 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entries 2QXV, 3LF9. The amino acid sequence of PDB entry 3LF9 was modified with the CCP4 program CHAINSAW to match the EB22 target sequence. Resolution: 2.55→45.5 Å / Cor.coef. Fo:Fc: 0.896 / Cor.coef. Fo:Fc free: 0.887 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.608 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.612 / SU Rfree Blow DPI: 0.282 / SU Rfree Cruickshank DPI: 0.285 Details: higher resolution EED structures, such as PDB entry 3K26, were used for the interpretation of poor electron density. EED backbone coordinates were restrained to a PHENIX.ROSETTA_REFINE model. ...Details: higher resolution EED structures, such as PDB entry 3K26, were used for the interpretation of poor electron density. EED backbone coordinates were restrained to a PHENIX.ROSETTA_REFINE model. Weak electron density at and around EB22 residue Leu-68 suggested a helical fold and was interpreted homologously to PDB entry 3LF9.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 139.43 Å2 / Biso mean: 53.36 Å2 / Biso min: 3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.36 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.55→45.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.55→2.73 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
