 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5v1s: Crystal structure of Streptococcus suis SuiB bound to S-adenosylm... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5v1s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Streptococcus suis SuiB bound to S-adenosylmethionine | ||||||
 Components Components | Radical SAM | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / Radical SAM enzyme / Lys-Trp crosslink / Streptococcus suis / metalloenzyme / SPASM domain / RRE domain | ||||||
| Function / homology |  Function and homology information Function and homology informationheme biosynthetic process / catalytic activity / 4 iron, 4 sulfur cluster binding / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Streptococcus suis (bacteria) Streptococcus suis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.492 Å | ||||||
 Authors Authors | Davis, K.M. / Bacik, J.P. / Ando, N. | ||||||
 Citation Citation | Journal: Proc. Natl. Acad. Sci. U.S.A. / Year: 2017 Title: Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition. Authors: Davis, K.M. / Schramma, K.R. / Hansen, W.A. / Bacik, J.P. / Khare, S.D. / Seyedsayamdost, M.R. / Ando, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5v1s.cif.gz | 184.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5v1s.ent.gz | 142.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5v1s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v1/5v1sftp://data.pdbj.org/pub/pdb/validation_reports/v1/5v1s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5v1qSC  5v1tC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53044.734 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus suis (bacteria) / Gene: ERS132399_01508 / Production host: Escherichia coli (E. coli) / References: UniProt: A0A0Z8EWX1#2: Chemical | ChemComp-SF4 / Iron–sulfur cluster  Mass: 351.640 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 351.640 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe4S4#3: Chemical | S-Adenosyl methionine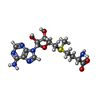  Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.58 % |
|---|---|
| Crystal grow | Temperature: 285 K / Method: vapor diffusion, sitting drop Details: A solution containing 18.9 mg/mL His6-tagged SuiB in in storage buffer [100 mM HEPES, pH 7.5, 300 mM KCl, 5 mM DTT, 10% (v/v) glycerol] was mixed 1:1 with precipitant solution. Small ...Details: A solution containing 18.9 mg/mL His6-tagged SuiB in in storage buffer [100 mM HEPES, pH 7.5, 300 mM KCl, 5 mM DTT, 10% (v/v) glycerol] was mixed 1:1 with precipitant solution. Small clusters of crystals were formed within 24 hrs. A seed stock was then produced by combining a single sitting well with 10 uL of reservoir solution and 10 uL of enzyme at 8 mg/mL, followed by brief vortexing. Following seeding, a resulting crystal was incubated in precipitant solution containing ~6 mM SAM for 30 min prior to transfer to cryosolution.The precipitant solution was 100 mM MES, pH 6.0, 15% (w/v) PEG 3350. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-D / Wavelength: 1.0332 Å / Beamline: 23-ID-D / Wavelength: 1.0332 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Feb 19, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0332 Å / Relative weight: 1 |
| Reflection | Resolution: 2.49→29.47 Å / Num. obs: 38407 / % possible obs: 99.8 % / Redundancy: 6.5 % / Rmerge(I) obs: 0.067 / Net I/σ(I): 17.7 |
| Reflection shell | Resolution: 2.49→2.59 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.839 / Mean I/σ(I) obs: 2 / Num. unique all: 4234 / % possible all: 99 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5V1Q Resolution: 2.492→29.47 Å / SU ML: 0.37 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 33.08
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.492→29.47 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
