 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5qcj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human Cathepsin-S with bound ligand | ||||||
 Components Components | Cathepsin S | ||||||
 Keywords Keywords | HYDROLASE / D3R / Cathepsin S / Ligand Docking | ||||||
| Function / homology |  Function and homology informationcathepsin S / basement membrane disassembly / positive regulation of cation channel activity / antigen processing and presentation of peptide antigen / endolysosome lumen / response to acidic pH / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures ...cathepsin S / basement membrane disassembly / positive regulation of cation channel activity / antigen processing and presentation of peptide antigen / endolysosome lumen / response to acidic pH / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures / toll-like receptor signaling pathway / cysteine-type endopeptidase activator activity involved in apoptotic process / fibronectin binding / antigen processing and presentation / collagen catabolic process / extracellular matrix disassembly / laminin binding / phagocytic vesicle / collagen binding / MHC class II antigen presentation / Degradation of the extracellular matrix / lysosomal lumen / proteolysis involved in protein catabolic process / positive regulation of apoptotic signaling pathway / Endosomal/Vacuolar pathway / protein processing / antigen processing and presentation of exogenous peptide antigen via MHC class II / late endosome / tertiary granule lumen / collagen-containing extracellular matrix / adaptive immune response / ficolin-1-rich granule lumen / lysosome / immune response / cysteine-type endopeptidase activity / serine-type endopeptidase activity / intracellular membrane-bounded organelle / Neutrophil degranulation / proteolysis / extracellular space / extracellular region Function and homology informationcathepsin S / basement membrane disassembly / positive regulation of cation channel activity / antigen processing and presentation of peptide antigen / endolysosome lumen / response to acidic pH / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures ...cathepsin S / basement membrane disassembly / positive regulation of cation channel activity / antigen processing and presentation of peptide antigen / endolysosome lumen / response to acidic pH / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures / toll-like receptor signaling pathway / cysteine-type endopeptidase activator activity involved in apoptotic process / fibronectin binding / antigen processing and presentation / collagen catabolic process / extracellular matrix disassembly / laminin binding / phagocytic vesicle / collagen binding / MHC class II antigen presentation / Degradation of the extracellular matrix / lysosomal lumen / proteolysis involved in protein catabolic process / positive regulation of apoptotic signaling pathway / Endosomal/Vacuolar pathway / protein processing / antigen processing and presentation of exogenous peptide antigen via MHC class II / late endosome / tertiary granule lumen / collagen-containing extracellular matrix / adaptive immune response / ficolin-1-rich granule lumen / lysosome / immune response / cysteine-type endopeptidase activity / serine-type endopeptidase activity / intracellular membrane-bounded organelle / Neutrophil degranulation / proteolysis / extracellular space / extracellular regionSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å | ||||||
| Model details | Structures re-refined for D3R docking challenge | ||||||
 Authors Authors | Bembenek, S.D. / Ameriks, M.K. / Mirzadegan, T. / Yang, H. / Shao, C. / Burley, S.K. | ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of human Cathepsin-S with bound ligand Authors: Bembenek, S.D. / Ameriks, M.K. / Mirzadegan, T. / Yang, H. / Shao, C. / Burley, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5qcj.cif.gz | 149.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5qcj.ent.gz | 123.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5qcj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qc/5qcjftp://data.pdbj.org/pub/pdb/validation_reports/qc/5qcj | HTTPS FTP |
|---|
-Group deposition
| ID | G_1002040 (26 entries) |
|---|---|
| Title | Ligand binding to Cathepsin S |
| Type | undefined |
| Description | Ligand binding to Cathepsin S |










-Related structure data
| Similar structure data |
|---|







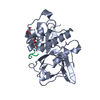


-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 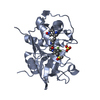
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
|
-Components
| #1: Protein | Mass: 24516.471 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSS / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P25774, cathepsin S Spodoptera frugiperda (fall armyworm) / References: UniProt: P25774, cathepsin S#2: Chemical |   Mass: 619.678 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C29H34F3N6O4S Mass: 619.678 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C29H34F3N6O4S#3: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 602 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 602 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.76 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 100MM SODIUM ACETATE PH 4.5, 200MM AMMONIUM ACETATE, 25% PEG 8000. PROTEIN CONCENTRATION 7 MG/ML, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.54178 |
| Detector | Type: RIGAKU SATURN 944 / Detector: CCD / Date: May 10, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 |
| Reflection | Resolution: 2→49.12 Å / Num. obs: 46139 / % possible obs: 95 % |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→49.12 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.938 / SU B: 4.472 / SU ML: 0.119 / Cross valid method: THROUGHOUT / ESU R: 0.194 / ESU R Free: 0.163 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.986 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2→49.12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|