 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
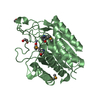
Yorodumi- PDB-5i9j: Structure of the cholesterol and lutein-binding domain of human S... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5i9j | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the cholesterol and lutein-binding domain of human STARD3 at 1.74A | ||||||
 Components Components | StAR-related lipid transfer protein 3 | ||||||
 Keywords Keywords |  TRANSPORT PROTEIN / lutein-binding protein / helix-grip fold / START / non-vesicular lipid transport TRANSPORT PROTEIN / lutein-binding protein / helix-grip fold / START / non-vesicular lipid transport | ||||||
| Function / homology |  Function and homology information Function and homology informationvesicle tethering to endoplasmic reticulum / progesterone biosynthetic process / endoplasmic reticulum-endosome membrane contact site / organelle membrane contact site / Pregnenolone biosynthesis / cholesterol transfer activity / cholesterol transport / cholesterol binding / mitochondrial transport / steroid metabolic process ...vesicle tethering to endoplasmic reticulum / progesterone biosynthetic process / endoplasmic reticulum-endosome membrane contact site / organelle membrane contact site / Pregnenolone biosynthesis / cholesterol transfer activity / cholesterol transport / cholesterol binding / mitochondrial transport / steroid metabolic process / cholesterol metabolic process / lipid metabolic process / late endosome membrane / endosome / lysosomal membrane / intracellular membrane-bounded organelle / endoplasmic reticulum membrane / protein homodimerization activity / mitochondrion / nucleoplasm / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.74 Å | ||||||
 Authors Authors | Horvath, M.P. / Bernstein, P.S. / Li, B. / George, E.W. / Tran, Q.T. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.F / Year: 2016 Title: Structure of the lutein-binding domain of human StARD3 at 1.74 angstrom resolution and model of a complex with lutein. Authors: Horvath, M.P. / George, E.W. / Tran, Q.T. / Baumgardner, K. / Zharov, G. / Lee, S. / Sharifzadeh, H. / Shihab, S. / Mattinson, T. / Li, B. / Bernstein, P.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5i9j.cif.gz | 104 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5i9j.ent.gz | 79.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5i9j.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i9/5i9jftp://data.pdbj.org/pub/pdb/validation_reports/i9/5i9j | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1em2S S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 25988.434 Da / Num. of mol.: 1 / Fragment: lutein-binding domain (UNP residues 216-444) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: STARD3, CAB1, MLN64 / Plasmid: pET22b(+)Details (production host): F- ompT hsdSB(rB- mB-) gal dcm lacY1 ahpC (DE3) gor522:: Tn10 trxB (KanR, TetR) Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): Origami B / References: UniProt: Q14849 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): Origami B / References: UniProt: Q14849 | ||
|---|---|---|---|
| #2: Chemical | ChemComp-TLA / Tartaric acid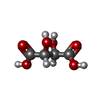  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 | ||
| #3: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 | ||
| #4: Chemical | Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 215 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 215 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 61 % / Description: hexagonal rod |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 9.1 Details: 0.85 M sodium/potassium tartrate, 0.2 M lithium sulfate, 0.1 M CHES, 0.01 M dithiothreitol. Cryostabilizer: 18% ethylene glycol, 5% ethanol in addition to 0.8M sodium/potassium tartrate, 0.1 ...Details: 0.85 M sodium/potassium tartrate, 0.2 M lithium sulfate, 0.1 M CHES, 0.01 M dithiothreitol. Cryostabilizer: 18% ethylene glycol, 5% ethanol in addition to 0.8M sodium/potassium tartrate, 0.1 M CHES pH 9.1, 0.2 M lithium sulfate, 0.01 M dithiothreitol. Crystals were frozen in propane and stored under liquid nitrogen Temp details: ambient |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 12.3.1 / Wavelength: 1.11583 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 19, 2015 |
| Radiation | Monochromator: DOUBLE CRYSTAL SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.11583 Å / Relative weight: 1 |
| Reflection | Resolution: 1.74→37.18 Å / Num. obs: 34160 / % possible obs: 99.5 % / Observed criterion σ(I): -3 / Redundancy: 8.8 % / CC1/2: 0.999 / Rmerge(I) obs: 0.046 / Net I/σ(I): 25.8 |
| Reflection shell | Resolution: 1.74→1.79 Å / Redundancy: 6.8 % / Rmerge(I) obs: 0.81 / Mean I/σ(I) obs: 2.4 / % possible all: 98.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1em2 Resolution: 1.74→37.18 Å / SU ML: 0.18 / σ(F): 1.34 / Phase error: 19.19 Details: riding hydrogens, 17 residues with alternate conformations,
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.5 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.74→37.18 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
