 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5hvp: CRYSTALLOGRAPHIC ANALYSIS OF A COMPLEX BETWEEN HUMAN IMMUNODEFICI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5hvp | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTALLOGRAPHIC ANALYSIS OF A COMPLEX BETWEEN HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 PROTEASE AND ACETYL-PEPSTATIN AT 2.0-ANGSTROMS RESOLUTION | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR /  ACID PROTEINASE / HYDROLASE-HYDROLASE INHIBITOR complex ACID PROTEINASE / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology informationHIV-1 retropepsin / : / retroviral ribonuclease H / exoribonuclease H / : / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA ...HIV-1 retropepsin / : / retroviral ribonuclease H / exoribonuclease H / : / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA / viral penetration into host nucleus / establishment of integrated proviral latency / RNA-directed DNA polymerase activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / RNA-DNA hybrid ribonuclease activity / viral nucleocapsid / endonuclease activity / DNA recombination / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / DNA-directed DNA polymerase activity / symbiont entry into host cell / symbiont-mediated suppression of host gene expression / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / RNA binding / zinc ion binding / membrane Function and homology informationHIV-1 retropepsin / : / retroviral ribonuclease H / exoribonuclease H / : / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA ...HIV-1 retropepsin / : / retroviral ribonuclease H / exoribonuclease H / : / exoribonuclease H activity / host multivesicular body / DNA integration / RNA-directed DNA polymerase / viral genome integration into host DNA / viral penetration into host nucleus / establishment of integrated proviral latency / RNA-directed DNA polymerase activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / RNA-DNA hybrid ribonuclease activity / viral nucleocapsid / endonuclease activity / DNA recombination / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / DNA-directed DNA polymerase activity / symbiont entry into host cell / symbiont-mediated suppression of host gene expression / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / RNA binding / zinc ion binding / membraneSimilarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 Streptomyces (bacteria) Streptomyces (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Fitzgerald, P.M.D. / Mckeever, B.M. / Vanmiddlesworth, J.F. / Springer, J.P. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1990 Title: Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0-A resolution. Authors: Fitzgerald, P.M. / McKeever, B.M. / VanMiddlesworth, J.F. / Springer, J.P. / Heimbach, J.C. / Leu, C.T. / Herber, W.K. / Dixon, R.A. / Darke, P.L. #1: Journal: Nature / Year: 1989Title: Three-Dimensional Structure of Aspartyl Protease from Human Immunodeficiency Virus HIV-1 Authors: Navia, M.A. / Fitzgerald, P.M.D. / Mckeever, B.M. / Leu, C.-T. / Heimbach, J.C. / Herber, W.K. / Sigal, I.S. / Darke, P.L. / Springer, J.P. #2: Journal: J.Biol.Chem. / Year: 1989Title: Crystallization of the Aspartylprotease from the Human Immunodefeciency Virus, HIV-1 Authors: Mckeever, B.M. / Navia, M.A. / Fitzgerald, P.M.D. / Springer, J.P. / Leu, C.-T. / Heimbach, J.C. / Herber, W.K. / Sigal, I.S. / Darke, P.L. #3: Journal: J.Biol.Chem. / Year: 1989Title: Human Immunodeficiency Virus Protease. Bacterial Expression and Characterization of the Purified Aspartic Protease Authors: Darke, P.L. / Leu, C.-T. / Davis, L.J. / Heimabch, J.C. / Diehl, R.E. / Hill, W.S. / Dixon, R.A.F. / Sigal, I.S. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET *S1* IS FORMED BY THE INTERDIGITATING STRANDS FROM THE TWO MONOMERS OF THE DIMERIC ...SHEET THE SHEET *S1* IS FORMED BY THE INTERDIGITATING STRANDS FROM THE TWO MONOMERS OF THE DIMERIC ENZYME. STRANDS 1 AND 3 ARE FROM CHAIN *A*. STRANDS 2 AND 4 ARE FROM CHAIN *B*. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5hvp.cif.gz | 54.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5hvp.ent.gz | 42.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5hvp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hv/5hvpftp://data.pdbj.org/pub/pdb/validation_reports/hv/5hvp | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









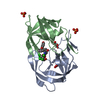


-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: SIDE CHAINS OF THESE RESIDUES HAVE BEEN MODELED IN TWO ALTERNATE CONFORMATIONS AND ASSIGNED ALTERNATE LOCATION INDICATORS *1* AND *2*. 2: THE OCCUPANCIES OF SOLVENT MOLECULES WHICH CORRELATED WITH STATIC DISORDER IN THE PROTEIN HAVE BEEN SET TO LESS THAN UNITY. WATER MOLECULES WITH ALTERNATE LOCATION INDICATOR *1* AND OCCUPANCY 0.58 ...2: THE OCCUPANCIES OF SOLVENT MOLECULES WHICH CORRELATED WITH STATIC DISORDER IN THE PROTEIN HAVE BEEN SET TO LESS THAN UNITY. WATER MOLECULES WITH ALTERNATE LOCATION INDICATOR *1* AND OCCUPANCY 0.58 CORRELATE WITH INHIBITOR ORIENTATIO *1*. WATER MOLECULES WITH ALTERNATE LOCATION INDICATOR *2* AND OCCUPANCY 0.42 CORRELATE WITH INHIBITOR ORIENTATION *2* IF THE SOLVENT MOLECULE IS DISTANT FROM THE BOUND INHIBITOR AN OCCUPANCY OF 0.50 HAS BEEN ASSIGNED. |
-Components
| #1: Protein | Mass: 10830.781 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Genus: Lentivirus / Production host: Escherichia coli (E. coli) / References: UniProt: Q9WFL7, UniProt: P12497*PLUS#2: Protein/peptide | | 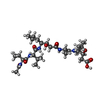  Type: Oligopeptide / Class: Inhibitor / Mass: 627.812 Da / Num. of mol.: 1 Type: Oligopeptide / Class: Inhibitor / Mass: 627.812 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces (bacteria) / References: ACETYL-PEPSTATIN#3: Chemical | Chloride  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2OCompound details | THE INHIBITOR BINDS TO THE ENZYME IN TWO ROUGHLY TWOFOLD SYMMETRIC ORIENTATIONS. THE ALTERNATE ...THE INHIBITOR BINDS TO THE ENZYME IN TWO ROUGHLY TWOFOLD SYMMETRIC ORIENTATIO | Sequence details | IN THE PAPER CITED IN THE *JRNL* RECORDS ABOVE, THE *B* CHAIN RESIDUES WERE NUMBERED FROM 201 TO ...IN THE PAPER CITED IN THE *JRNL* RECORDS ABOVE, THE *B* CHAIN RESIDUES WERE NUMBERED FROM 201 TO 299. THIS NUMBERING SCHEME HAS BEEN RETAINED. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.63 Å3/Da / Density % sol: 53.16 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 5.4 / Method: vapor diffusion / Details: macro-seeding | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.92 Å / Lowest resolution: 9999 Å / Num. obs: 13397 / % possible obs: 72.6 % / Observed criterion σ(I): 1 / Num. measured all: 36470 / Rmerge(I) obs: 0.0439 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2→8 Å / σ(I): 0 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 8 Å / Num. reflection obs: 12901 / Rfactor obs: 0.176 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
