 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5hll: Re-refinement of 4g4a: room-temperature X-ray diffraction study o... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5hll | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Re-refinement of 4g4a: room-temperature X-ray diffraction study of cisplatin and its binding to His15 of HEWL after 14 months chemical exposure in the presence of DMSO. | |||||||||
 Components Components | Lysozyme C | |||||||||
 Keywords Keywords |  HYDROLASE / cisplatin / histidine / DMSO / Hen egg white lysozyme / raw diffraction images data HYDROLASE / cisplatin / histidine / DMSO / Hen egg white lysozyme / raw diffraction images data | |||||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium / defense response to bacterium / endoplasmic reticulum / extracellular space / identical protein binding / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Gallus gallus (chicken) Gallus gallus (chicken) | |||||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | |||||||||
 Authors Authors | Helliwell, J.R. | |||||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| |||||||||
 Citation Citation | Journal: Acta Crystallogr F Struct Biol Commun / Year: 2016 Title: Re-refinement of 4g4a: room-temperature X-ray diffraction study of cisplatin and its binding to His15 of HEWL after 14 months chemical exposure in the presence of DMSO. Authors: Tanley, S.W. / Schreurs, A.M. / Kroon-Batenburg, L.M. / Helliwell, J.R. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2012 Title: Towards automated crystallographic structure refinement with phenix.refine. Authors: Afonine, P.V. / Grosse-Kunstleve, R.W. / Echols, N. / Headd, J.J. / Moriarty, N.W. / Mustyakimov, M. / Terwilliger, T.C. / Urzhumtsev, A. / Zwart, P.H. / Adams, P.D. #2: Journal: Acta Crystallogr D Biol Crystallogr / Year: 2010 Title: PHENIX: a comprehensive Python-based system for macromolecular structure solution. Authors: Paul D Adams / Pavel V Afonine / Gábor Bunkóczi / Vincent B Chen / Ian W Davis / Nathaniel Echols / Jeffrey J Headd / Li-Wei Hung / Gary J Kapral / Ralf W Grosse-Kunstleve / Airlie J McCoy ...Authors: Paul D Adams / Pavel V Afonine / Gábor Bunkóczi / Vincent B Chen / Ian W Davis / Nathaniel Echols / Jeffrey J Headd / Li-Wei Hung / Gary J Kapral / Ralf W Grosse-Kunstleve / Airlie J McCoy / Nigel W Moriarty / Robert Oeffner / Randy J Read / David C Richardson / Jane S Richardson / Thomas C Terwilliger / Peter H Zwart /  Abstract: Macromolecular X-ray crystallography is routinely applied to understand biological processes at a molecular level. However, significant time and effort are still required to solve and complete many ...Macromolecular X-ray crystallography is routinely applied to understand biological processes at a molecular level. However, significant time and effort are still required to solve and complete many of these structures because of the need for manual interpretation of complex numerical data using many software packages and the repeated use of interactive three-dimensional graphics. PHENIX has been developed to provide a comprehensive system for macromolecular crystallographic structure solution with an emphasis on the automation of all procedures. This has relied on the development of algorithms that minimize or eliminate subjective input, the development of algorithms that automate procedures that are traditionally performed by hand and, finally, the development of a framework that allows a tight integration between the algorithms. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5hll.cif.gz | 76 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5hll.ent.gz | 46.3 KB | Display | PDB format |
| PDBx/mmJSON format | 5hll.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hl/5hllftp://data.pdbj.org/pub/pdb/validation_reports/hl/5hll | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4g4a S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Experimental dataset #1 | Data reference: 10.15127/1.215887 / Data set type: diffraction image data |

















-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
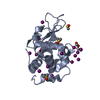
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: Hen egg white lyysozyme with cisplatin with DMSO / Source: (natural) Gallus gallus (chicken) / Tissue: egg white / References: UniProt: P00698, lysozyme |
|---|
-Non-polymers , 6 types, 43 molecules 




| #2: Chemical | ChemComp-DMS / Dimethyl sulfoxide Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM | ||||||||
|---|---|---|---|---|---|---|---|---|---|
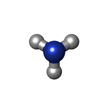
| #3: Chemical | ChemComp-CL / Chloride Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#5: Chemical |  Mass: 195.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Pt Mass: 195.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Pt#6: Chemical | Ammonia Mass: 17.031 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: NH3 Mass: 17.031 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: NH3#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.75 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: batch mode / pH: 4.7 Details: HEWL CO-CRYSTALLIZED WITH CISPLATIN, WITH 5% DMSO MEDIA IN 1 ML, 10% SODIUM CHLORIDE + 1 ML 0.04 M SODIUM ACETATE, PH 4.7, BATCH, TEMPERATURE 295K |
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.54 Å |
| Detector | Type: APEX II CCD / Detector: CCD / Date: Jan 25, 2012 |
| Radiation | Monochromator: CONFOCAL MIRROR OPTICS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.699→35.39 Å / Num. obs: 13804 / % possible obs: 99.68 % / Redundancy: 6.7 % / Biso Wilson estimate: 18.9861835344 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.048 / Net I/σ(I): 9.8 |
| Reflection shell | Resolution: 1.699→1.728 Å / Redundancy: 4.5 % / Rmerge(I) obs: 2.428 / Mean I/σ(I) obs: 0.56 / % possible all: 93.86 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4g4a 4g4a Resolution: 1.7→35.39 Å / SU ML: 0.220931737624 / SU R Cruickshank DPI: 0.177 / Cross valid method: FREE R-VALUE / Phase error: 23.7110864462
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.3947691261 Å2 | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→35.39 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
