 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ftg: Human choline kinase a1 in complex with compound 1-[[4-[2-[4-[[4-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ftg | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human choline kinase a1 in complex with compound 1-[[4-[2-[4-[[4-(dimethylamino)pyridin-1- yl]methyl]phenoxy]ethoxy]phenyl]methyl]-N,N- dimethyl-pyridin-4-amine (compound 10a) | |||||||||
 Components Components | CHOLINE KINASE ALPHA | |||||||||
 Keywords Keywords |  TRANSFERASE / BISCATIONIC INHIBITOR / DOCKING STUDIES TRANSFERASE / BISCATIONIC INHIBITOR / DOCKING STUDIES | |||||||||
| Function / homology |  Function and homology informationethanolamine kinase / choline kinase / ethanolamine kinase activity / CDP-choline pathway / choline kinase activity / Synthesis of PE / phosphatidylethanolamine biosynthetic process / lipid droplet disassembly / phosphatidylcholine biosynthetic process / cholinesterase activity ...ethanolamine kinase / choline kinase / ethanolamine kinase activity / CDP-choline pathway / choline kinase activity / Synthesis of PE / phosphatidylethanolamine biosynthetic process / lipid droplet disassembly / phosphatidylcholine biosynthetic process / cholinesterase activity / lipid transport / Synthesis of PC / cellular response to glucose starvation / lipid droplet / lipid metabolic process / protein tyrosine kinase activity / phosphorylation / protein homodimerization activity / ATP binding / cytosol / cytoplasm Function and homology informationethanolamine kinase / choline kinase / ethanolamine kinase activity / CDP-choline pathway / choline kinase activity / Synthesis of PE / phosphatidylethanolamine biosynthetic process / lipid droplet disassembly / phosphatidylcholine biosynthetic process / cholinesterase activity ...ethanolamine kinase / choline kinase / ethanolamine kinase activity / CDP-choline pathway / choline kinase activity / Synthesis of PE / phosphatidylethanolamine biosynthetic process / lipid droplet disassembly / phosphatidylcholine biosynthetic process / cholinesterase activity / lipid transport / Synthesis of PC / cellular response to glucose starvation / lipid droplet / lipid metabolic process / protein tyrosine kinase activity / phosphorylation / protein homodimerization activity / ATP binding / cytosol / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | |||||||||
 Authors Authors | Schiaffino-Ortega, S. / Baglioni, E. / Mariotto, E. / Bortolozzi, R. / Serran-Aguilera, L. / Rios-Marco, P. / Carrasco-Jimenez, M.P. / Gallo, M.A. / Hurtado-Guerrero, R. / Marco, C. ...Schiaffino-Ortega, S. / Baglioni, E. / Mariotto, E. / Bortolozzi, R. / Serran-Aguilera, L. / Rios-Marco, P. / Carrasco-Jimenez, M.P. / Gallo, M.A. / Hurtado-Guerrero, R. / Marco, C. / Basso, G. / Viola, G. / Entrena, A. / Lopez-Cara, L.C. | |||||||||
 Citation Citation | Journal: Sci.Rep. / Year: 2016 Title: Design, Synthesis, Crystallization and Biological Evaluation of New Symmetrical Biscationic Compounds as Selective Inhibitors of Human Choline Kinase Alpha1 (Chokalpha1) Authors: Schiaffino-Ortega, S. / Baglioni, E. / Mariotto, E. / Bortolozzi, R. / Serran-Aguilera, L. / Rios-Marco, P. / Carrasco-Jimenez, M.P. / Gallo, M.A. / Hurtado-Guerrero, R. / Marco, C. / Basso, ...Authors: Schiaffino-Ortega, S. / Baglioni, E. / Mariotto, E. / Bortolozzi, R. / Serran-Aguilera, L. / Rios-Marco, P. / Carrasco-Jimenez, M.P. / Gallo, M.A. / Hurtado-Guerrero, R. / Marco, C. / Basso, G. / Viola, G. / Entrena, A. / Lopez-Cara, L.C. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ftg.cif.gz | 160.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ftg.ent.gz | 126 KB | Display | PDB format |
| PDBx/mmJSON format | 5ftg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ft/5ftgftp://data.pdbj.org/pub/pdb/validation_reports/ft/5ftg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3g15S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 44463.902 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 80-457 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PMALC2X / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): STAR ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): STARReferences: UniProt: P35790, choline kinase, ethanolamine kinase | ||
|---|---|---|---|
| #2: Chemical | ChemComp-NBR / 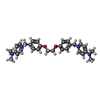  Mass: 484.632 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H36N4O2 Mass: 484.632 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H36N4O2 | ||
| #3: Chemical | ChemComp-EDO / Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C2H6O2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 298 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 298 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.03 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 1.0507 / Beamline: I04-1 / Wavelength: 1.0507 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0507 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→58 Å / Num. obs: 74167 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 23.2 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 34.8 |
| Reflection shell | Resolution: 1.45→1.53 Å / Redundancy: 14 % / Rmerge(I) obs: 0.67 / Mean I/σ(I) obs: 5 / % possible all: 99.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3G15 Resolution: 1.45→58.48 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.951 / SU B: 1.931 / SU ML: 0.037 / Cross valid method: THROUGHOUT / ESU R: 0.06 / ESU R Free: 0.063 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.966 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→58.48 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|