 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5fpd: Structure of heat shock-related 70kDA protein 2 with small-molecu... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5fpd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of heat shock-related 70kDA protein 2 with small-molecule ligand pyrazine-2-carboxamide (AT513) in an alternate binding site. | ||||||
 Components Components | HEAT SHOCK-RELATED 70KDA PROTEIN 2 | ||||||
 Keywords Keywords |  CHAPERONE / HEAT SHOCK-RELATED PROTEIN / HEAT SHOCK / HSP70 / HSPA2 / PROTEIN-LIGAND COMPLEX / FRAGMENT SCREENING / ALTERNATE BINDING SITE / AT513. CHAPERONE / HEAT SHOCK-RELATED PROTEIN / HEAT SHOCK / HSP70 / HSPA2 / PROTEIN-LIGAND COMPLEX / FRAGMENT SCREENING / ALTERNATE BINDING SITE / AT513. | ||||||
| Function / homology |  Function and homology informationCatSper complex / glycolipid binding / synaptonemal complex disassembly / negative regulation of inclusion body assembly / male meiotic nuclear division / synaptonemal complex / meiotic spindle / male meiosis I / chaperone cofactor-dependent protein refolding / spermatid development ...CatSper complex / glycolipid binding / synaptonemal complex disassembly / negative regulation of inclusion body assembly / male meiotic nuclear division / synaptonemal complex / meiotic spindle / male meiosis I / chaperone cofactor-dependent protein refolding / spermatid development / response to unfolded protein / Regulation of HSF1-mediated heat shock response / Attenuation phase / protein folding chaperone / heat shock protein binding / positive regulation of G2/M transition of mitotic cell cycle / Meiotic synapsis / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / response to cold / male germ cell nucleus / ATP-dependent protein folding chaperone / PKR-mediated signaling / tau protein binding / disordered domain specific binding / unfolded protein binding / protein-folding chaperone binding / response to heat / protein refolding / spermatogenesis / blood microparticle / positive regulation of protein phosphorylation / enzyme binding / cell surface / ATP hydrolysis activity / extracellular exosome / ATP binding / membrane / nucleus / plasma membrane / cytosol / cytoplasm Function and homology informationCatSper complex / glycolipid binding / synaptonemal complex disassembly / negative regulation of inclusion body assembly / male meiotic nuclear division / synaptonemal complex / meiotic spindle / male meiosis I / chaperone cofactor-dependent protein refolding / spermatid development ...CatSper complex / glycolipid binding / synaptonemal complex disassembly / negative regulation of inclusion body assembly / male meiotic nuclear division / synaptonemal complex / meiotic spindle / male meiosis I / chaperone cofactor-dependent protein refolding / spermatid development / response to unfolded protein / Regulation of HSF1-mediated heat shock response / Attenuation phase / protein folding chaperone / heat shock protein binding / positive regulation of G2/M transition of mitotic cell cycle / Meiotic synapsis / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / response to cold / male germ cell nucleus / ATP-dependent protein folding chaperone / PKR-mediated signaling / tau protein binding / disordered domain specific binding / unfolded protein binding / protein-folding chaperone binding / response to heat / protein refolding / spermatogenesis / blood microparticle / positive regulation of protein phosphorylation / enzyme binding / cell surface / ATP hydrolysis activity / extracellular exosome / ATP binding / membrane / nucleus / plasma membrane / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.97 Å | ||||||
 Authors Authors | Jhoti, H. / Ludlow, R.F. / Patel, S. / Saini, H.K. / Tickle, I.J. / Verdonk, M. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2015 Title: Detection of Secondary Binding Sites in Proteins Using Fragment Screening. Authors: Ludlow, R.F. / Verdonk, M.L. / Saini, H.K. / Tickle, I.J. / Jhoti, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5fpd.cif.gz | 310.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5fpd.ent.gz | 252.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5fpd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fp/5fpdftp://data.pdbj.org/pub/pdb/validation_reports/fp/5fpd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5fp5C  5fp6C  5fpeC  5fpmC  5fpnC  5fpoC  5fprC  5fpsC  5fptC  5fpyC  1s3xS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (1, 0.00025, 0.00087), Vector : |
-Components
| #1: Protein | Mass: 42383.000 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PET28B / Production host:  ESCHERICHIA COLI BL21(DE3) (bacteria) / References: UniProt: P54652 ESCHERICHIA COLI BL21(DE3) (bacteria) / References: UniProt: P54652#2: Chemical | Pyrazinamide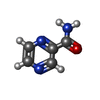  Mass: 123.113 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H5N3O / Comment: medication*YM Mass: 123.113 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H5N3O / Comment: medication*YM#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 878 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 878 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | PYRAZINE-2-CARBOXAMIDE (L02): ASTEX COMPOUND REGISTRY AT513. PYRAZINE-2-CARBOXAMIDE (L01): ASTEX ...PYRAZINE-2-CARBOXAMID | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.3 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8 Details: 1.0M NACL, 0.1M TRIS/HCL PH=8, 20.0% W/V PEG 8000. PROTEIN CONC. = 9MG/ML. |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E+ / Wavelength: 1.5418 |
| Detector | Type: RIGAKU SATURN CCD / Detector: CCD / Date: May 30, 2008 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.97→38.3 Å / Num. obs: 53188 / % possible obs: 97.9 % / Observed criterion σ(I): -3.7 / Redundancy: 2 % / Biso Wilson estimate: 22.79 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 12.7 |
| Reflection shell | Resolution: 1.97→2.03 Å / Rmerge(I) obs: 0.16 / Mean I/σ(I) obs: 3.7 / % possible all: 88.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1S3X Resolution: 1.97→38.34 Å / Cor.coef. Fo:Fc: 0.9504 / Cor.coef. Fo:Fc free: 0.9179 / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.45 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.97→38.34 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.97→2.02 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
