 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5d9g: Crystal structure of TIPRL, TOR signaling pathway regulator-like,... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5d9g | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of TIPRL, TOR signaling pathway regulator-like, in complex with peptide | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords |  PROTEIN BINDING / protein binding TIP41-like family PP2A PROTEIN BINDING / protein binding TIP41-like family PP2A | ||||||||||||
| Function / homology |  Function and homology informationregulation of phosphoprotein phosphatase activity / negative regulation of phosphoprotein phosphatase activity / TOR signaling / DNA damage checkpoint signaling / cytosol Function and homology informationregulation of phosphoprotein phosphatase activity / negative regulation of phosphoprotein phosphatase activity / TOR signaling / DNA damage checkpoint signaling / cytosolSimilarity search - Function | ||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / SAD / Resolution: 2.15 Å | ||||||||||||
 Authors Authors | Scorsato, V. / Sandy, J. / Brandao-Neto, J. / Pereira, H.M. / Smetana, J.H.C. / Aparicio, R. | ||||||||||||
| Funding support |  Brazil, 3items Brazil, 3items
| ||||||||||||
 Citation Citation | Journal: Sci Rep / Year: 2016 Title: Crystal structure of the human Tip41 orthologue, TIPRL, reveals a novel fold and a binding site for the PP2Ac C-terminus. Authors: Scorsato, V. / Lima, T.B. / Righetto, G.L. / Zanchin, N.I. / Brandao-Neto, J. / Sandy, J. / Pereira, H.D. / Ferrari, A.J. / Gozzo, F.C. / Smetana, J.H. / Aparicio, R. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5d9g.cif.gz | 221 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5d9g.ent.gz | 186.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5d9g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d9/5d9gftp://data.pdbj.org/pub/pdb/validation_reports/d9/5d9g | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 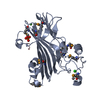
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE BIOLOGICAL UNIT HAS BEEN DETERMINED BY SAXS AND GEL FILTRATION |
-Components
-Protein / Protein/peptide , 2 types, 4 molecules ABCD
| #1: Protein | Mass: 28843.502 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TIPRL / Production host:  Escherichia coli (E. coli) / References: UniProt: O75663 Escherichia coli (E. coli) / References: UniProt: O75663#2: Protein/peptide | Mass: 812.866 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host: Escherichia coli (E. coli) |
|---|
-Non-polymers , 4 types, 238 molecules 



| #3: Chemical | Chloride Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Chemical | Phosphate Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#5: Chemical | ChemComp-NI / | Nickel Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.95 Å3/Da / Density % sol: 75.15 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 5.5 / Details: 100mM SPG buffer, pH 5.5. 1.5M NaCl, TIPRL 90mg/mL / PH range: 5.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.99219 Å / Beamline: I02 / Wavelength: 0.99219 Å | ||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Dec 9, 2014 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.99219 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 2.15→95.95 Å / Num. obs: 62695 / % possible obs: 100 % / Redundancy: 15.2 % / Biso Wilson estimate: 44.69 Å2 / CC1/2: 0.995 / Rmerge(I) obs: 0.119 / Rpim(I) all: 0.031 / Net I/σ(I): 12.1 / Num. measured all: 952365 / Scaling rejects: 241 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0 / % possible all: 100
|
-Phasing
| Phasing | Method: SAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.15→52.616 Å / SU ML: 0.3 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 27.89 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 163.94 Å2 / Biso mean: 58.5944 Å2 / Biso min: 29.84 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.15→52.616 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 18
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
