 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ca9: Structures of the candida albicans sey1p GTPase in complex with G... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ca9 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structures of the candida albicans sey1p GTPase in complex with GDPAlF4- | ||||||||||||
 Components Components | Protein SEY1 | ||||||||||||
 Keywords Keywords |  HYDROLASE / ER / Sey1p / membrance fusion / dynamin HYDROLASE / ER / Sey1p / membrance fusion / dynamin | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationhexose transmembrane transport / endoplasmic reticulum inheritance / endoplasmic reticulum membrane fusion / cortical endoplasmic reticulum / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / GTPase activity / endoplasmic reticulum membrane / GTP binding / endoplasmic reticulumSimilarity search - Function | ||||||||||||
| Biological species |  Candida albicans (yeast) Candida albicans (yeast) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.8 Å | ||||||||||||
 Authors Authors | Yan, L. | ||||||||||||
| Funding support |  China, 3items China, 3items
| ||||||||||||
 Citation Citation | Journal: J.Cell Biol. / Year: 2015 Title: Structures of the yeast dynamin-like GTPase Sey1p provide insight into homotypic ER fusion Authors: Yan, L. / Sun, S. / Wang, W. / Shi, J. / Hu, X. / Wang, S. / Su, D. / Rao, Z. / Hu, J. / Lou, Z. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ca9.cif.gz | 275.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ca9.ent.gz | 233 KB | Display | PDB format |
| PDBx/mmJSON format | 5ca9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ca/5ca9ftp://data.pdbj.org/pub/pdb/validation_reports/ca/5ca9 | HTTPS FTP |
|---|
-Related structure data








-Links
 PDBj
PDBj




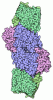

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 79124.000 Da / Num. of mol.: 1 / Fragment: UNP residues 1-692 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Candida albicans (strain SC5314 / ATCC MYA-2876) (yeast)Strain: SC5314 / ATCC MYA-2876 / Gene: SEY1, NAG6, CaO19.2151, CaO19.9698 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q9C0L9, dynamin GTPase Escherichia coli (E. coli) / References: UniProt: Q9C0L9, dynamin GTPase |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-ALF / 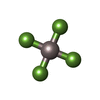  Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4 Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4 |
| #4: Chemical | ChemComp-GDP / Guanosine diphosphate  Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | 1. SEQUENCE CONFLICTS D270G, A337T AND I479V ARE BASED ON BAB43823.1 ACCORDING TO DATABASE Q9C0L9 ...1. SEQUENCE CONFLICTS D270G, A337T AND I479V ARE BASED ON BAB43823.1 ACCORDING TO DATABASE Q9C0L9 (SEY1_CANAL). 2. THE CODON CUG (MRNA CUG CORRESPOND |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.47 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: evaporation Details: 160 mM Lithium sulfate monohydrate, 80 mM Tris pH 8.5, 20% w/v Polyethylene glycol 3,350, 40 mM Ammonium acetate, 20 mM Sodium citrate tribasic dihydrate pH 5.6, 6% w/v Polyethylene glycol ...Details: 160 mM Lithium sulfate monohydrate, 80 mM Tris pH 8.5, 20% w/v Polyethylene glycol 3,350, 40 mM Ammonium acetate, 20 mM Sodium citrate tribasic dihydrate pH 5.6, 6% w/v Polyethylene glycol 4,000, 20 mM spermidine |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Dec 8, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→50 Å / Num. obs: 21774 / % possible obs: 99 % / Redundancy: 10.28 % / Net I/σ(I): 43.9 |
| Reflection shell | Resolution: 2.8→2.85 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→48.76 Å / Cor.coef. Fo:Fc: 0.911 / Cor.coef. Fo:Fc free: 0.902 / SU B: 30.112 / SU ML: 0.316 / Cross valid method: THROUGHOUT / ESU R Free: 0.41 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 78.318 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→48.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|