 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4zbg: Crystal Structure of a GNAT family Acetyltransferase from Brucell... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zbg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of a GNAT family Acetyltransferase from Brucella melitensis in complex with Acetyl-CoA | ||||||
 Components Components | Acetyltransferase | ||||||
 Keywords Keywords | TRANSFERASE / SGCID / N-acetyltransferase / Acyl_CoA_acyltransferase / Structural Genomics / Seattle Structural Genomics Center for Infectious Disease / SSGCID | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Brucella abortus str. 2308 A (bacteria) Brucella abortus str. 2308 A (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.25 Å | ||||||
 Authors Authors | Seattle Structural Genomics Center for Infectious Disease (SSGCID) | ||||||
 Citation Citation | Journal: to be published Title: Crystal Structure of a GNAT family Acetyltransferase from Brucella melitensis in complex with Acetyl-CoA Authors: SSGCID / Dranow, D.M. / Trakaki, T. / Lorimer, D. / Edwards, T.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zbg.cif.gz | 87.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zbg.ent.gz | 62 KB | Display | PDB format |
| PDBx/mmJSON format | 4zbg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zb/4zbgftp://data.pdbj.org/pub/pdb/validation_reports/zb/4zbg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4rs2S S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18631.240 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Brucella abortus str. 2308 A (bacteria)Strain: 2308 A / Gene: BAAA_2000484 / Plasmid: BrabA.17468.b.B1 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: C4IR59 |
|---|---|
| #2: Chemical | ChemComp-ACO / Acetyl-CoA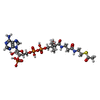  Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S |
| #3: Chemical | ChemComp-GOL / Glycerol  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 241 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 241 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38.5 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: BrabA.17468.b.B1.PS02320 at 10.4mg/ml, mixed with 4mM acetyl-CoA, then 1:1 with MCSG1(c12): 0.1M Bis-Tris:HCl, pH=6.5, 25% PEG-3350, cryo-protected with 20% ethylene glycol |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-F / Wavelength: 0.97872 Å / Beamline: 21-ID-F / Wavelength: 0.97872 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Mar 12, 2015 / Details: Beryllium Lenses | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Diamond [111] / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97872 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Number: 218334 / Rmerge(I) obs: 0.04 / Χ2: 0.99 / D res high: 1.25 Å / Num. obs: 40267 / % possible obs: 99.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.25→50 Å / Num. obs: 40267 / % possible obs: 99.1 % / Observed criterion σ(I): -3 / Redundancy: 5.4 % / Biso Wilson estimate: 9.23 Å2 / Rmerge F obs: 0.999 / Rmerge(I) obs: 0.04 / Rrim(I) all: 0.045 / Χ2: 0.991 / Net I/σ(I): 23.66 / Num. measured all: 218334 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4RS2 Resolution: 1.25→26.291 Å / SU ML: 0.1 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 15.92 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 62.45 Å2 / Biso mean: 15.191 Å2 / Biso min: 4.7 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.25→26.291 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
|
