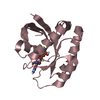
A: Tetrapyrrole-binding protein B: Tetrapyrrole-binding protein C: Tetrapyrrole-binding protein D: Tetrapyrrole-binding protein E: Tetrapyrrole-binding protein F: Tetrapyrrole-binding protein
Mass: 25823.041 Da / Num. of mol.: 6 / Fragment: residues 40-260 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Chlamydomonas reinhardtii (plant) / Gene: GUN4, CHLREDRAFT_205768 / Production host: Escherichia coli (E. coli) / References: UniProt: A8I5N5
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION
-
Sample preparation
Crystal
Density Matthews: 2.64 Å3/Da / Density % sol: 53 % / Description: very small hexagonal rods
Crystal grow
Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 1:1 ratio of protein (35 mg/mL in 20 mM Tricine (pH 8.0), 2 mM beta-mercaptoethanol) to well solution (1.0 M ammonium citrate (pH 7.0), 0.1 M Bis Tris Propane (pH 7.0). Crystals only ...Details: 1:1 ratio of protein (35 mg/mL in 20 mM Tricine (pH 8.0), 2 mM beta-mercaptoethanol) to well solution (1.0 M ammonium citrate (pH 7.0), 0.1 M Bis Tris Propane (pH 7.0). Crystals only appeared after 44 weeks, likely the time taken for proteolysis, resulting in the N- and C-terminal truncations PH range: 7.0-8.0
-
Data collection
Diffraction
Mean temperature: 100 K
Diffraction source
Source: SYNCHROTRON / Site: Australian Synchrotron / Beamline: MX2 / Wavelength: 0.9537 Å
Resolution: 3.5→44.5 Å / Cor.coef. Fo:Fc: 0.879 / Cor.coef. Fo:Fc free: 0.813 / SU B: 43.036 / SU ML: 0.663 / Cross valid method: THROUGHOUT / ESU R Free: 0.77 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.31315
753
5.4 %
RANDOM
Rwork
0.25602
-
-
-
obs
0.25907
13187
99.05 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords PLANT PROTEIN
PLANT PROTEIN Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj Assembly
Assembly







 Sample preparation
Sample preparation / Beamline: MX2 / Wavelength: 0.9537 Å
/ Beamline: MX2 / Wavelength: 0.9537 Å Processing
Processing