 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xa7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Soluble part of holo NqrC from V. harveyi | ||||||
 Components Components | Na(+)-translocating NADH-quinone reductase subunit C | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / Na+-translocating NADH:quinone oxidoreductase / redox-driven sodium pump OXIDOREDUCTASE / Na+-translocating NADH:quinone oxidoreductase / redox-driven sodium pump | ||||||
| Function / homology | FLAVIN MONONUCLEOTIDE / :  Function and homology information Function and homology information | ||||||
| Biological species |  Vibrio harveyi CAIM 1792 (bacteria) Vibrio harveyi CAIM 1792 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.56 Å | ||||||
 Authors Authors | Borshchevskiy, V. / Round, E. / Bertsova, Y. / Polovinkin, V. / Gushchin, I. / Mishin, A. / Kovalev, K. / Kachalova, G. / Popov, A. / Bogachev, A. / Gordeliy, V. | ||||||
 Citation Citation | Journal: Plos One / Year: 2015 Title: Structural and Functional Investigation of Flavin Binding Center of the NqrC Subunit of Sodium-Translocating NADH:Quinone Oxidoreductase from Vibrio harveyi. Authors: Borshchevskiy, V. / Round, E. / Bertsova, Y. / Polovinkin, V. / Gushchin, I. / Ishchenko, A. / Kovalev, K. / Mishin, A. / Kachalova, G. / Popov, A. / Bogachev, A. / Gordeliy, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xa7.cif.gz | 106.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xa7.ent.gz | 80.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4xa7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xa/4xa7ftp://data.pdbj.org/pub/pdb/validation_reports/xa/4xa7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3lwxS S: Starting model for refinement |
|---|---|
| Similar structure data |






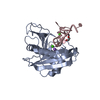



-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27553.756 Da / Num. of mol.: 1 / Fragment: UNP residues 33-261 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vibrio harveyi CAIM 1792 (bacteria) / Gene: nqrC, MUQ_18848 / Production host: Escherichia coli (E. coli)References: UniProt: M7R347, Oxidoreductases; Acting on NADH or NADPH; With a quinone or similar compound as acceptor |
|---|---|
| #2: Chemical | ChemComp-FMN / Flavin mononucleotide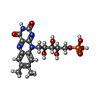  Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P |
| #3: Chemical | ChemComp-CL / Chloride  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 113 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 113 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.85 Å3/Da / Density % sol: 33.54 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / Details: 0.1 M K thiocyanate, 20-30%(w/v) PEG MME 2000 / PH range: 6.8 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.976 Å / Beamline: ID29 / Wavelength: 0.976 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Mar 17, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976 Å / Relative weight: 1 |
| Reflection | Resolution: 1.56→46.53 Å / Num. all: 28742 / Num. obs: 27755 / % possible obs: 96.6 % / Observed criterion σ(F): -3 / Redundancy: 4.1 % / Biso Wilson estimate: 31.8 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 9.6 |
| Reflection shell | Resolution: 1.56→1.6 Å / Redundancy: 3.3 % / Rmerge(I) obs: 1.62 / Mean I/σ(I) obs: 0.63 / % possible all: 74.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3LWX Resolution: 1.56→46.53 Å / SU ML: 0.26 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 33.06 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.56→46.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
