 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4x32: Bacteriorhodopsin ground state structure collected in cryo condit... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4x32 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bacteriorhodopsin ground state structure collected in cryo conditions from crystals obtained in LCP with PEG as a precipitant. | ||||||
 Components Components | Bacteriorhodopsin | ||||||
 Keywords Keywords | PROTON TRANSPORT / light-driven proton pump / retinal binding / seven transmembrane helix protein | ||||||
| Function / homology |  Function and homology informationphotoreceptor activity / phototransduction / proton transmembrane transport / monoatomic ion channel activity / plasma membrane Function and homology informationphotoreceptor activity / phototransduction / proton transmembrane transport / monoatomic ion channel activity / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Halobacterium salinarum (Halophile) Halobacterium salinarum (Halophile) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å | ||||||
 Authors Authors | Nogly, P. / Standfuss, J. | ||||||
 Citation Citation | Journal: Iucrj / Year: 2015 Title: Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. Authors: Nogly, P. / James, D. / Wang, D. / White, T.A. / Zatsepin, N. / Shilova, A. / Nelson, G. / Liu, H. / Johansson, L. / Heymann, M. / Jaeger, K. / Metz, M. / Wickstrand, C. / Wu, W. / Bath, P. ...Authors: Nogly, P. / James, D. / Wang, D. / White, T.A. / Zatsepin, N. / Shilova, A. / Nelson, G. / Liu, H. / Johansson, L. / Heymann, M. / Jaeger, K. / Metz, M. / Wickstrand, C. / Wu, W. / Bath, P. / Berntsen, P. / Oberthuer, D. / Panneels, V. / Cherezov, V. / Chapman, H. / Schertler, G. / Neutze, R. / Spence, J. / Moraes, I. / Burghammer, M. / Standfuss, J. / Weierstall, U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4x32.cif.gz | 109.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4x32.ent.gz | 82.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4x32.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/x3/4x32ftp://data.pdbj.org/pub/pdb/validation_reports/x3/4x32 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4x31C  2ntuS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / BR / Bacterioopsin / BO Mass: 24990.535 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 18-245 / Source method: isolated from a natural source / Source: (natural) Halobacterium salinarum (Halophile) / Strain: ATCC 700922 / JCM 11081 / NRC-1 / Tissue: purple membrane / References: UniProt: P02945 | ||
|---|---|---|---|
| #2: Chemical | ChemComp-RET / Retinal  Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O | ||
| #3: Chemical | ChemComp-LI1 / 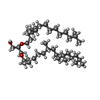  Mass: 639.130 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C42H86O3 Mass: 639.130 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C42H86O3#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.71 % Description: hexagonal plate with 50 microns in the longest dimension |
|---|---|
| Crystal grow | Temperature: 294 K / Method: lipidic cubic phase / pH: 5.6 / Details: 27% PEG2000, 100 mM phosphate buffer pH 5.6 / PH range: 5.4-5.8 |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: cryostream of LN2 | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Feb 15, 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Number: 274871 / Rmerge(I) obs: 0.107 / Χ2: 1.06 / D res high: 1.79 Å / Num. obs: 19786 / % possible obs: 99.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.9→46.57 Å / Num. all: 16647 / Num. obs: 16643 / % possible obs: 100 % / Redundancy: 14.1 % / Biso Wilson estimate: 33.4 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.095 / Rpim(I) all: 0.026 / Net I/σ(I): 17.9 / Num. measured all: 234541 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2NTU Resolution: 1.9→52.42 Å / Cor.coef. Fo:Fc: 0.964 / Cor.coef. Fo:Fc free: 0.939 / SU B: 7.283 / SU ML: 0.094 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.158 / ESU R Free: 0.143 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 93.4 Å2 / Biso mean: 36.741 Å2 / Biso min: 22.33 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.9→52.42 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 85.0075 Å / Origin y: 32.9867 Å / Origin z: -4.5054 Å
|
