 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4tyq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of an adenylate kinase mutant--AKm2 | ||||||
 Components Components | Adenylate kinase | ||||||
 Keywords Keywords | TRANSFERASE / ATP binding | ||||||
| Function / homology |  Function and homology information Function and homology informationnucleoside monophosphate metabolic process / nucleoside diphosphate metabolic process / adenylate kinase / adenylate kinase activity / AMP salvage / nucleoside diphosphate kinase activity / phosphorylation / zinc ion binding / ATP binding / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Bacillus subtilis 168 (bacteria) Bacillus subtilis 168 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Moon, S. / Bae, E. | ||||||
 Citation Citation | Journal: J KOREAN SOC APPL BIOL CHEM / Year: 2014 Title: Crystal structures of thermally stable adenylate kinase mutants designed by local structural entropy optimization and structure-guided mutagenesis Authors: Moon, S. / Bae, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4tyq.cif.gz | 112.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4tyq.ent.gz | 83.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4tyq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ty/4tyqftp://data.pdbj.org/pub/pdb/validation_reports/ty/4tyq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4typC  1zioS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | / AK / ATP-AMP transphosphorylase / ATP:AMP phosphotransferase / Adenylate monophosphate kinase / ...AK / ATP-AMP transphosphorylase / ATP:AMP phosphotransferase / Adenylate monophosphate kinase / Superoxide-inducible protein 16 / SOI16 Mass: 24406.096 Da / Num. of mol.: 2 Mutation: L3I, G17A, E22A, D23K, K69R, G73S, D75S, Y103M, K105R, P106K, I107L, D108E, N112H, D106R, K107Q, D108E, V109E, S169A, T179M, Q180A, D184A, S187D, E188S, G190E, Y191V, A193V, I201M, Q202E, ...Mutation: L3I, G17A, E22A, D23K, K69R, G73S, D75S, Y103M, K105R, P106K, I107L, D108E, N112H, D106R, K107Q, D108E, V109E, S169A, T179M, Q180A, D184A, S187D, E188S, G190E, Y191V, A193V, I201M, Q202E, D203K, Y205F, A206K, V208L, K209R, D210E, L211I, G213Q, K216A, K217R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus subtilis 168 (bacteria) / Gene: adk, BSU01370 / Plasmid: pET11a / Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: P16304, adenylate kinase |
|---|
-Non-polymers , 5 types, 339 molecules 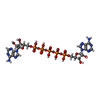




| #2: Chemical |  Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5#3: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#5: Chemical | ChemComp-CA / |  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 332 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.66 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop Details: 22% (w/v) polyethylene glycol 3350, 200mM calcium chloride |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS  / Beamline: 5C (4A) / Wavelength: 0.9796 Å / Beamline: 5C (4A) / Wavelength: 0.9796 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: May 1, 2014 |
| Radiation | Monochromator: DCM Si (111) Crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9796 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→50 Å / Num. all: 47801 / Num. obs: 45746 / % possible obs: 95.7 % / Redundancy: 1.9 % / Rmerge(I) obs: 0.074 / Net I/σ(I): 8.4 |
| Reflection shell | Resolution: 1.65→1.71 Å / Redundancy: 1.9 % / Rmerge(I) obs: 0.198 / Mean I/σ(I) obs: 4.6 / % possible all: 92.3 |
- Processing
Processing
| Software | Name: REFMAC / Version: 5.8.0049 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ZIO Resolution: 1.65→50 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.934 / SU B: 4.167 / SU ML: 0.07 / Cross valid method: THROUGHOUT / ESU R: 0.11 / ESU R Free: 0.111 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.729 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.65→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|