 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4o7d | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human glutaminase in complex DON | ||||||
 Components Components | Glutaminase kidney isoform, mitochondrial | ||||||
 Keywords Keywords |  HYDROLASE HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationglutamine catabolic process / regulation of respiratory gaseous exchange by nervous system process / glutamate biosynthetic process / Glutamate and glutamine metabolism / intracellular glutamate homeostasis / Glutamate Neurotransmitter Release Cycle / glutaminase / glutaminase activity / suckling behavior / TP53 Regulates Metabolic Genes ...glutamine catabolic process / regulation of respiratory gaseous exchange by nervous system process / glutamate biosynthetic process / Glutamate and glutamine metabolism / intracellular glutamate homeostasis / Glutamate Neurotransmitter Release Cycle / glutaminase / glutaminase activity / suckling behavior / TP53 Regulates Metabolic Genes / chemical synaptic transmission / protein homotetramerization / mitochondrial matrix / synapse / mitochondrion / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Thangavelu, K. / Sivaraman, J. | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2014 Title: Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA) Authors: Thangavelu, K. / Chong, Q.Y. / Low, B.C. / Sivaraman, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4o7d.cif.gz | 71 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4o7d.ent.gz | 56.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4o7d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o7/4o7dftp://data.pdbj.org/pub/pdb/validation_reports/o7/4o7d | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 34461.355 Da / Num. of mol.: 1 / Fragment: UNP Residues 221-531 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GLS, GLS1, KIAA0838 / Production host:  Escherichia coli (E. coli) / References: UniProt: O94925, glutaminase Escherichia coli (E. coli) / References: UniProt: O94925, glutaminase |
|---|---|
| #2: Chemical | ChemComp-ONL / 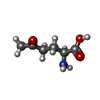  Type: L-peptide linking / Mass: 145.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H11NO3 Type: L-peptide linking / Mass: 145.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H11NO3 |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.470732 Å3/Da / Density % sol: 77.516716 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.2 Details: 0.1M Bis-Tris propane (pH 7.2), 1.8M Lithium Sulphate, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12B / Beamline: X12B |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.3→30 Å / Num. obs: 34102 / % possible obs: 99.6 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.3→25.771 Å / FOM work R set: 0.8508 / SU ML: 0.61 / σ(F): 1.42 / Phase error: 21.86 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.95 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 33.353 Å2 / ksol: 0.342 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 150.74 Å2 / Biso mean: 54.57 Å2 / Biso min: 29.59 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→25.771 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 12
|
