Entry Database : PDB / ID : 4nzgTitle Crystal Structure of the N-terminal domain of Moloney murine leukemia virus integrase, Northeast Structural Genomics Consortium Target OR3 Integrase p46 Keywords / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Method / / / Resolution : 2.152 Å Authors Guan, R. / Jiang, M. / Janjua, H. / Maglaqui, M. / Zhao, L. / Xiao, R. / Acton, T.B. / Everett, J.K. / Roth, M. / Montelione, G.T. / Northeast Structural Genomics Consortium (NESG) Journal : Proteins / Year : 2017Title : X-ray crystal structure of the N-terminal region of Moloney murine leukemia virus integrase and its implications for viral DNA recognition.Authors : Guan, R. / Aiyer, S. / Cote, M.L. / Xiao, R. / Jiang, M. / Acton, T.B. / Roth, M.J. / Montelione, G.T. History Deposition Dec 12, 2013 Deposition site / Processing site Revision 1.0 Feb 5, 2014 Provider / Type Revision 1.1 Feb 22, 2017 Group Revision 1.2 Aug 24, 2022 Group / Derived calculationsCategory citation / database_2 ... citation / database_2 / pdbx_struct_conn_angle / struct_conn / struct_ref_seq_dif / struct_site Item _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_asym_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_label_asym_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.3 Sep 20, 2023 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords VIRAL PROTEIN /
VIRAL PROTEIN /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj




 Assembly
Assembly




 Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn
 Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2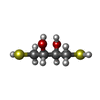
 Mass: 154.251 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C4H10O2S2
Mass: 154.251 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C4H10O2S2 Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X29A / Wavelength: 1.075 Å
/ Beamline: X29A / Wavelength: 1.075 Å Processing
Processing