 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4kru: X-ray structure of catalytic domain of endolysin from clostridium... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4kru | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | X-ray structure of catalytic domain of endolysin from clostridium perfringens phage phiSM101 | |||||||||
 Components Components | Autolytic lysozyme | |||||||||
 Keywords Keywords |  HYDROLASE / beta/alpha barrel / muramidase HYDROLASE / beta/alpha barrel / muramidase | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activitySimilarity search - Function | |||||||||
| Biological species |  Clostridium phage phiSM101 (virus) Clostridium phage phiSM101 (virus) | |||||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.37 Å | |||||||||
 Authors Authors | Kamitori, S. / Yoshida, H. | |||||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2014 Title: X-ray structure of a novel endolysin encoded by episomal phage phiSM101 of Clostridium perfringens. Authors: Tamai, E. / Yoshida, H. / Sekiya, H. / Nariya, H. / Miyata, S. / Okabe, A. / Kuwahara, T. / Maki, J. / Kamitori, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4kru.cif.gz | 65.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4kru.ent.gz | 46.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4kru.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kr/4kruftp://data.pdbj.org/pub/pdb/validation_reports/kr/4kru | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4krtC  1jfxS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25894.516 Da / Num. of mol.: 1 / Fragment: catalytic domain, UNP residues 1-218 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Clostridium phage phiSM101 (virus) / Strain: SM101 / Gene: CPR_C0050 / Plasmid: pCold II / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21-CodonPlus-RIL / References: UniProt: Q0SPG7, lysozyme Escherichia coli (E. coli) / Strain (production host): BL21-CodonPlus-RIL / References: UniProt: Q0SPG7, lysozyme |
|---|---|
| #2: Sugar | ChemComp-NDG / N-Acetylglucosamine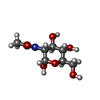  Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
| #3: Sugar | ChemComp-NAG / N-Acetylglucosamine  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
| #4: Chemical | ChemComp-PO4 / Phosphate  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 346 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 346 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.95 Å3/Da / Density % sol: 36.96 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 4.2 Details: 20% PEG 3000, 0.2M Lithium sulphate, 0.1M phosphate-citrate, pH 4.2, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS VII / Detector: IMAGE PLATE / Date: Sep 27, 2012 / Details: mirrors (VariMax Optic) |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.37→30.96 Å / Num. all: 42675 / Num. obs: 42675 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 6.15 % / Biso Wilson estimate: 10.9 Å2 / Rmerge(I) obs: 0.024 / Net I/σ(I): 42.5 |
| Reflection shell | Resolution: 1.37→1.42 Å / Redundancy: 2.52 % / Rmerge(I) obs: 0.071 / Mean I/σ(I) obs: 11.7 / Num. unique all: 3633 / % possible all: 85 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1JFX Resolution: 1.37→30.96 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 1163041.84 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 49.7347 Å2 / ksol: 0.4 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.9 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.37→30.96 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.37→1.42 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
