 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hp3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Tet3 in complex with a CpG dsDNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA BINDING PROTEIN/DNA / CXXC /  DNA methylation / DNA BINDING PROTEIN-DNA complex DNA methylation / DNA BINDING PROTEIN-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationmethylcytosine dioxygenase / 5-methylcytosine catabolic process / 5-methylcytosine dioxygenase activity / methyl-CpG binding / DNA demethylation / chromosome / chromatin organization / regulation of gene expression / zinc ion binding / nucleusSimilarity search - Function | ||||||
| Biological species | XENOPUS TROPICALIS | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.05 Å | ||||||
 Authors Authors | Chao, X. / Tempel, W. / Bian, C. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2012 Title: Tet3 CXXC Domain and Dioxygenase Activity Cooperatively Regulate Key Genes for Xenopus Eye and Neural Development. Authors: Xu, Y. / Xu, C. / Kato, A. / Tempel, W. / Abreu, J.G. / Bian, C. / Hu, Y. / Hu, D. / Zhao, B. / Cerovina, T. / Diao, J. / Wu, F. / He, H.H. / Cui, Q. / Clark, E. / Ma, C. / Barbara, A. / ...Authors: Xu, Y. / Xu, C. / Kato, A. / Tempel, W. / Abreu, J.G. / Bian, C. / Hu, Y. / Hu, D. / Zhao, B. / Cerovina, T. / Diao, J. / Wu, F. / He, H.H. / Cui, Q. / Clark, E. / Ma, C. / Barbara, A. / Veenstra, G.J. / Xu, G. / Kaiser, U.B. / Liu, X.S. / Sugrue, S.P. / He, X. / Min, J. / Kato, Y. / Shi, Y.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hp3.cif.gz | 63.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hp3.ent.gz | 43.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4hp3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hp/4hp3ftp://data.pdbj.org/pub/pdb/validation_reports/hp/4hp3 | HTTPS FTP |
|---|
-Related structure data


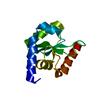








-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 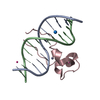
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | AUTHORS STATE THAT THE BIOLOGICAL ASSEMBLY IS UNKNOWN. |
-Components
| #1: DNA chain | Mass: 3663.392 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: Protein | | Mass: 8430.049 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) XENOPUS (SILURANA) TROPICALIS (tropical clawed frog)Plasmid: pET28-MHL / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21-V2R-pRARE2 / References: UniProt: A0JP82 Escherichia coli (E. coli) / Strain (production host): BL21-V2R-pRARE2 / References: UniProt: A0JP82#3: Chemical |   Num. of mol.: 3 / Source method: obtained synthetically Num. of mol.: 3 / Source method: obtained synthetically#4: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.33 Å3/Da / Density % sol: 47.28 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 30% PEG-1500, 0.2M sodium chloride, 0.1M HEPES, pH 7.5, vapor diffusion, sitting drop, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.97949 Å / Beamline: 08ID-1 / Wavelength: 0.97949 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jan 25, 2012 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97949 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.05→34.42 Å / Num. obs: 9281 / % possible obs: 99.79 % / Redundancy: 3.71 % / Rmerge(I) obs: 0.053 / Net I/σ(I): 12.5727 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: unpublished model of complex between same protein construct and a methylated DNA fragment. That structure was solved by a combination of zinc-SAD (SHELXD/E) and molecular replacement ...Starting model: unpublished model of complex between same protein construct and a methylated DNA fragment. That structure was solved by a combination of zinc-SAD (SHELXD/E) and molecular replacement in "density" mode (MOLREP), using ideal DNA (COOT) and human MLL2 (currently unpublished) as search models. Resolution: 2.05→30 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.948 / WRfactor Rfree: 0.256 / WRfactor Rwork: 0.22 / Occupancy max: 1 / Occupancy min: 0.5 / SU B: 8.214 / SU ML: 0.139 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.186 / ESU R Free: 0.162 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED. COOT and the MOLPROBITY server were also used during refinement.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 122.83 Å2 / Biso mean: 52.769 Å2 / Biso min: 27.08 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
