Journal: PLoS Pathog / Year: 2014 Title: The CD27L and CTP1L endolysins targeting Clostridia contain a built-in trigger and release factor. Authors: Matthew Dunne / Haydyn D T Mertens / Vasiliki Garefalaki / Cy M Jeffries / Andrew Thompson / Edward A Lemke / Dmitri I Svergun / Melinda J Mayer / Arjan Narbad / Rob Meijers / Abstract: The bacteriophage ΦCD27 is capable of lysing Clostridium difficile, a pathogenic bacterium that is a major cause for nosocomial infection. A recombinant CD27L endolysin lyses C. difficile in vitro, ...The bacteriophage ΦCD27 is capable of lysing Clostridium difficile, a pathogenic bacterium that is a major cause for nosocomial infection. A recombinant CD27L endolysin lyses C. difficile in vitro, and represents a promising alternative as a bactericide. To better understand the lysis mechanism, we have determined the crystal structure of an autoproteolytic fragment of the CD27L endolysin. The structure covers the C-terminal domain of the endolysin, and represents a novel fold that is identified in a number of lysins that target Clostridia bacteria. The structure indicates endolysin cleavage occurs at the stem of the linker connecting the catalytic domain with the C-terminal domain. We also solved the crystal structure of the C-terminal domain of a slow cleaving mutant of the CTP1L endolysin that targets C. tyrobutyricum. Two distinct dimerization modes are observed in the crystal structures for both endolysins, despite a sequence identity of only 22% between the domains. The dimers are validated to be present for the full length protein in solution by right angle light scattering, small angle X-ray scattering and cross-linking experiments using the cross-linking amino acid p-benzoyl-L-phenylalanine (pBpa). Mutagenesis on residues contributing to the dimer interfaces indicates that there is a link between the dimerization modes and the autocleavage mechanism. We show that for the CTP1L endolysin, there is a reduction in lysis efficiency that is proportional to the cleavage efficiency. We propose a model for endolysin triggering, where the extended dimer presents the inactive state, and a switch to the side-by-side dimer triggers the cleavage of the C-terminal domain. This leads to the release of the catalytic portion of the endolysin, enabling the efficient digestion of the bacterial cell wall.
Mass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O
Sequence details
VAL195 HAS BEEN MUTATED TO A PROLINE TO REDUCE AUTOCLEAVAGE
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.4 Å3/Da / Density % sol: 48 % Description: THERE ARE ICE RINGS, AND THE DETECTOR GEOMETRY LIMITED HIGH RESOLUTION. BEAMLINE WAS UNDER COMMISSIONING.
Crystal grow
pH: 8 / Details: 20MM TRIS PH8.0 AND 6 % PEG 8000
-
Data collection
Diffraction
Mean temperature: 100 K
Diffraction source
Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY / Beamline: P14 (MX2) / Wavelength: 1.223
Detector
Type: MARMOSAIC 225 mm CCD / Detector: CCD / Details: DIRECT BEAM (NO MIRRORS)
Radiation
Monochromator: SI 1 1 1 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.223 Å / Relative weight: 1
Reflection
Resolution: 2.1→20 Å / Num. obs: 4489 / % possible obs: 92 % / Observed criterion σ(I): 0 / Redundancy: 5.7 % / Rmerge(I) obs: 0.02 / Net I/σ(I): 48.9
Reflection shell
Highest resolution: 2.1 Å
-
Processing
Software
Name
Version
Classification
REFMAC
5.7.0029
refinement
DENZO
datareduction
SCALEPACK
datascaling
MOLREP
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: CD27L C-TERMINAL DOMAIN Resolution: 2.11→16.54 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.9 / SU B: 5.669 / SU ML: 0.143 / Cross valid method: THROUGHOUT / ESU R: 0.262 / ESU R Free: 0.236 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.26382
221
4.7 %
RANDOM
Rwork
0.17217
-
-
-
obs
0.17617
4489
91.03 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Lysin
Lysin  Keywords
Keywords Function and homology information
Function and homology information Clostridium phage phiCTP1 (virus)
Clostridium phage phiCTP1 (virus) Authors
Authors Citation
Citation


 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj Assembly
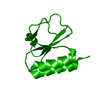
Assembly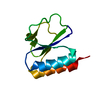

 Mass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing