 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4bum: Crystal structure of the Voltage Dependant Anion Channel 2 from z... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4bum | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the Voltage Dependant Anion Channel 2 from zebrafish. | ||||||
 Components Components | VOLTAGE-DEPENDENT ANION CHANNEL 2 | ||||||
 Keywords Keywords |  MEMBRANE PROTEIN / MITOCHONDRIA / PORIN / MEMBRANE / DETERGENT / RECOMBINANT MEMBRANE PROTEIN / MITOCHONDRIA / PORIN / MEMBRANE / DETERGENT / RECOMBINANT | ||||||
| Function / homology |  Function and homology information Function and homology informationfin regeneration / Ub-specific processing proteases / voltage-gated monoatomic anion channel activity / calcium import into the mitochondrion / regulation of heart contraction / channel activity / porin activity / pore complex / mitochondrial outer membrane / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  DANIO RERIO (zebrafish) DANIO RERIO (zebrafish) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.801 Å | ||||||
 Authors Authors | Paz, A. / Schredelseker, J. / Abramson, J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2014 Title: High-Resolution Structure and Double Electron-Electron Resonance of the Zebrafish Voltage Dependent Anion Channel 2 Reveal an Oligomeric Population. Authors: Schredelseker, J. / Paz, A. / Lopez, C.J. / Altenbach, C. / Leung, C.S. / Drexler, M.K. / Chen, J. / Hubbell, W.L. / Abramson, J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "XA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "XA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 19-STRANDED BARREL THIS IS REPRESENTED BY A 20-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4bum.cif.gz | 121.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4bum.ent.gz | 94.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4bum.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bu/4bumftp://data.pdbj.org/pub/pdb/validation_reports/bu/4bum | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3emnS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31148.822 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) DANIO RERIO (zebrafish) / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): M15 / References: UniProt: Q8AWD0 ESCHERICHIA COLI (E. coli) / Strain (production host): M15 / References: UniProt: Q8AWD0 |
|---|---|
| #2: Chemical | ChemComp-LDA / Lauryldimethylamine oxide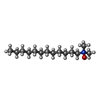  Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8 Details: 100 MM TRISHCL (PH=8.0), 100 MM KCL, AND PEG 2000 IN A RANGE OF 19-24%, 0.011% N-UNDECYL-BETA-D-THIOMALTOPYRANOSIDE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1 / Beamline: 5.0.2 / Wavelength: 1 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 16, 2012 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→51.29 Å / Num. obs: 13686 / % possible obs: 98 % / Observed criterion σ(I): 2 / Redundancy: 3.56 % / Biso Wilson estimate: 70.86 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 13 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3EMN Resolution: 2.801→51.286 Å / SU ML: 0.37 / σ(F): 2 / Phase error: 27.48 / Stereochemistry target values: ML Details: SOME RESIDUES WERE TRIMMED DUE TO VERY WEAK ELECTRON DENSITY OF SIDE-CHAINS. THE LOOP BETWEEN RESIDUES 266-272 IS VERY MOBILE. WE ARE CERTAIN OF THE GENERAL LOCATION IN WHICH IT IS MODELLED, ...Details: SOME RESIDUES WERE TRIMMED DUE TO VERY WEAK ELECTRON DENSITY OF SIDE-CHAINS. THE LOOP BETWEEN RESIDUES 266-272 IS VERY MOBILE. WE ARE CERTAIN OF THE GENERAL LOCATION IN WHICH IT IS MODELLED, BUT IN SOLUTION IT COULD SAMPLE MANY POSITIONS AROUND THIS VICINITY.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 50.95 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.801→51.286 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
