Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9184 Å / Relative weight: 1
Reflection
Resolution: 2.2→35 Å / Num. obs: 339176 / % possible obs: 99.6 % / Observed criterion σ(I): 2 / Redundancy: 13.6 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 16
Reflection shell
Resolution: 2.2→2.26 Å / Redundancy: 4.1 % / Rmerge(I) obs: 0.51 / Mean I/σ(I) obs: 2.7 / % possible all: 99.7
-
Processing
Software
Name
Version
Classification
PHENIX
(PHENIX.REFINE: 1.7_650)
refinement
AutoSol
phasing
Refinement
Method to determine structure: SIRAS Starting model: NONE Resolution: 2.2→19.936 Å / SU ML: 0.29 / σ(F): 1.99 / Phase error: 27.76 / Stereochemistry target values: ML Details: RESIDUES FROM 3 TO 107 IN CHAIN WERE NOT MODELED BECAUSE OF NO OBSERVABLE ELECTRON DENSITY. RESIDUES CYS38 TO CYS44 IN CHAIN X WERE MODELED STEREOCHEMICALLY.
Rfactor
Num. reflection
% reflection
Rfree
0.258
4174
5 %
Rwork
0.22
-
-
obs
0.222
83461
99.8 %
Solvent computation
Shrinkage radii: 0.61 Å / VDW probe radii: 0.9 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 55.258 Å2 / ksol: 0.366 e/Å3
Displacement parameters
Baniso -1
Baniso -2
Baniso -3
1-
16.8817 Å2
0 Å2
0 Å2
2-
-
-7.5969 Å2
0 Å2
3-
-
-
-9.2848 Å2
Refinement step
Cycle: LAST / Resolution: 2.2→19.936 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
8781
0
79
396
9256
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.007
9378
X-RAY DIFFRACTION
f_angle_d
1.836
12321
X-RAY DIFFRACTION
f_dihedral_angle_d
16.476
3382
X-RAY DIFFRACTION
f_chiral_restr
0.052
1435
X-RAY DIFFRACTION
f_plane_restr
0.003
1580
Refine LS restraints NCS
Ens-ID
Dom-ID
Auth asym-ID
Number
Refine-ID
Type
Rms dev position (Å)
1
1
X
3897
X-RAY DIFFRACTION
POSITIONAL
1
2
Y
3897
X-RAY DIFFRACTION
POSITIONAL
0.083
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.2-2.225
0.3642
139
0.3133
2627
X-RAY DIFFRACTION
100
2.225-2.2511
0.3645
134
0.3005
2567
X-RAY DIFFRACTION
100
2.2511-2.2785
0.3191
141
0.299
2670
X-RAY DIFFRACTION
100
2.2785-2.3073
0.3106
134
0.2764
2551
X-RAY DIFFRACTION
100
2.3073-2.3376
0.3249
139
0.2712
2633
X-RAY DIFFRACTION
100
2.3376-2.3696
0.3297
137
0.2598
2597
X-RAY DIFFRACTION
100
2.3696-2.4034
0.2892
139
0.2676
2644
X-RAY DIFFRACTION
100
2.4034-2.4392
0.3106
136
0.2662
2590
X-RAY DIFFRACTION
100
2.4392-2.4773
0.351
139
0.2711
2633
X-RAY DIFFRACTION
100
2.4773-2.5178
0.3772
137
0.2831
2617
X-RAY DIFFRACTION
100
2.5178-2.5611
0.3055
137
0.2781
2595
X-RAY DIFFRACTION
100
2.5611-2.6076
0.3405
139
0.2609
2641
X-RAY DIFFRACTION
100
2.6076-2.6577
0.3058
136
0.2567
2590
X-RAY DIFFRACTION
100
2.6577-2.7118
0.2723
141
0.2515
2660
X-RAY DIFFRACTION
100
2.7118-2.7706
0.3008
136
0.2478
2587
X-RAY DIFFRACTION
100
2.7706-2.8349
0.3008
139
0.2511
2649
X-RAY DIFFRACTION
100
2.8349-2.9055
0.2806
137
0.255
2607
X-RAY DIFFRACTION
100
2.9055-2.9839
0.2755
139
0.2445
2641
X-RAY DIFFRACTION
100
2.9839-3.0714
0.2693
139
0.2422
2640
X-RAY DIFFRACTION
100
3.0714-3.1701
0.261
140
0.2444
2653
X-RAY DIFFRACTION
100
3.1701-3.2829
0.2762
138
0.2316
2634
X-RAY DIFFRACTION
100
3.2829-3.4138
0.2896
140
0.2252
2654
X-RAY DIFFRACTION
100
3.4138-3.5683
0.2775
139
0.2179
2640
X-RAY DIFFRACTION
100
3.5683-3.7552
0.2557
141
0.2004
2672
X-RAY DIFFRACTION
100
3.7552-3.9888
0.2245
140
0.1953
2662
X-RAY DIFFRACTION
100
3.9888-4.2939
0.2053
139
0.177
2651
X-RAY DIFFRACTION
100
4.2939-4.7208
0.2125
142
0.1646
2695
X-RAY DIFFRACTION
100
4.7208-5.3921
0.2297
142
0.1902
2702
X-RAY DIFFRACTION
100
5.3921-6.7494
0.2459
145
0.2164
2747
X-RAY DIFFRACTION
100
6.7494-19.9371
0.1801
150
0.1841
2838
X-RAY DIFFRACTION
99
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords ATPASE
ATPASE Function and homology information
Function and homology information

 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj








 Assembly
Assembly
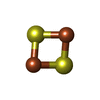



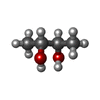

 Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2
Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4 Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2
Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2 Sample preparation
Sample preparation / Beamline: 14.2 / Wavelength: 0.9184
/ Beamline: 14.2 / Wavelength: 0.9184  Processing
Processing