 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3rdi: Domain-domain flexibility leads to allostery within the camp rece... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rdi | ||||||
|---|---|---|---|---|---|---|---|
| Title | Domain-domain flexibility leads to allostery within the camp receptor protein (CRP) | ||||||
 Components Components | Catabolite gene activator | ||||||
 Keywords Keywords |  TRANSCRIPTION REGULATOR / CAMP RECEPTOR PROTEIN (CRP) / ALLOSTERY / DNA BINDING CYCLIC AMP / CATABOLITE ACTIVATOR PROTEIN (CAP) / DNA BINDING PROTEIN TRANSCRIPTION REGULATOR / CAMP RECEPTOR PROTEIN (CRP) / ALLOSTERY / DNA BINDING CYCLIC AMP / CATABOLITE ACTIVATOR PROTEIN (CAP) / DNA BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationcarbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / DNA-templated transcription / positive regulation of DNA-templated transcription ...carbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / DNA-templated transcription / positive regulation of DNA-templated transcription / identical protein binding / cytosolSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.95 Å | ||||||
 Authors Authors | Knapp, J. / White, M.A. / Lee, J.C. | ||||||
 Citation Citation | Journal: To be Published Title: Domain-Domain Flexibility Leads to Allostery within the Cam Receptor Protein (Crp) Authors: Knapp, J. / White, M.A. / Lee, J.C. #1: Journal: J.Mol.Biol. / Year: 1987 Title: Structure of a Complex of Catabolite Gene Activator Protein and Cyclic AMP at 2.5 A Resolution Authors: Weber, I.T. / Steitz, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rdi.cif.gz | 91.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rdi.ent.gz | 69.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3rdi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rd/3rdiftp://data.pdbj.org/pub/pdb/validation_reports/rd/3rdi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3rouC  3rpqC  3rypC  3ryrC  1il5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
| #1: Protein | Mass: 23732.537 Da / Num. of mol.: 2 / Mutation: S63F Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Strain: K12 / Gene: b3357, cap, crp, csm, JW5702 / Production host: ESCHERICHIA COLI (E. coli) / References: UniProt: P0ACJ8#2: Chemical | Cyclic adenosine monophosphate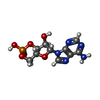  Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.51 Å3/Da / Density % sol: 50.96 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 15% ETHANOL, 100MM TRIS, 3MM CAMP, PH 7.00, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 298K |
-Data collection
| Diffraction | Mean temperature: 85 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: MACSCIENCE / Wavelength: 1.5418 |
| Detector | Type: MAC Science DIP-2030 / Detector: IMAGE PLATE / Date: Nov 9, 2007 / Details: CONFOCAL |
| Radiation | Monochromator: MULTILAYER OPTICS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.95→40 Å / Num. all: 9807 / Num. obs: 9807 / % possible obs: 98.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 4.7 % / Biso Wilson estimate: 134.9 Å2 / Rmerge(I) obs: 0.068 / Rsym value: 0.068 / Net I/σ(I): 20.6 |
| Reflection shell | Resolution: 2.95→3.06 Å / Redundancy: 4.6 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 2.8 / Rsym value: 0.45 / % possible all: 96.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1IL5 Resolution: 2.95→34.65 Å / Rfactor Rfree error: 0.013 / Data cutoff high absF: 1670547.31 / Data cutoff low absF: 0 / Isotropic thermal model: variable / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber / Details: 2 NCS GROUPS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 74.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.95→34.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Rms dev Biso : 0 Å2 / Rms dev position: 0 Å / Weight Biso : 2 / Weight position: 2000
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.95→3 Å / Rfactor Rfree error: 0.087 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
