 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
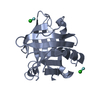
Yorodumi- PDB-3ph5: Bovine beta lactoglobulin crystallized through ligandation of ytt... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3ph5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bovine beta lactoglobulin crystallized through ligandation of yttrium cations | ||||||
 Components Components | Beta-lactoglobulin | ||||||
 Keywords Keywords | TRANSPORT PROTEIN | ||||||
| Function / homology |  Function and homology informationretinol binding / long-chain fatty acid binding / extracellular space / identical protein binding Function and homology informationretinol binding / long-chain fatty acid binding / extracellular space / identical protein bindingSimilarity search - Function | ||||||
| Biological species |  Bos taurus (cattle) Bos taurus (cattle) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.4 Å | ||||||
 Authors Authors | Zocher, G. / Stehle, T. | ||||||
 Citation Citation | Journal: J.Appl.Crystallogr. / Year: 2011 Title: Novel Approach to Protein Crystallization through Ligandation of Yttrium Cations Authors: Zhang, F. / Zocher, G. / Sauter, A. / Stehle, T. / Schreiber, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3ph5.cif.gz | 69.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3ph5.ent.gz | 55.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3ph5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ph/3ph5ftp://data.pdbj.org/pub/pdb/validation_reports/ph/3ph5 | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / Beta-LG Mass: 18188.018 Da / Num. of mol.: 2 / Fragment: unp residues 18-178 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bos taurus (cattle) / References: UniProt: P02754#2: Chemical | ChemComp-YT3 / Yttrium  Mass: 88.906 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Y Mass: 88.906 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Y#3: Chemical | Chloride  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 48.52 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: batch crystallization / pH: 7 Details: Crystallized in presents of 3.0 mM yttrium(III) chlorid, pH 7, Batch Crystallization, temperature 277K |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 2.4→20 Å / Num. all: 14221 / Num. obs: 14221 / % possible obs: 99.8 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 3 / Redundancy: 5.5 % / Biso Wilson estimate: 42.3 Å2 / Rmerge(I) obs: 0.111 / Rsym value: 0.068 / Net I/σ(I): 19.1 | ||||||||||||||||||
| Reflection shell | Resolution: 2.4→2.46 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.662 / Mean I/σ(I) obs: 3 / Num. unique all: 1930 / Rsym value: 0.523 / % possible all: 99.9 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.4→19.59 Å / Cor.coef. Fo:Fc: 0.933 / Cor.coef. Fo:Fc free: 0.916 / SU B: 20.819 / SU ML: 0.216 / Cross valid method: THROUGHOUT / ESU R Free: 0.276 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.233 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→19.59 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.461 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
