 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3p53 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of fascin | ||||||
 Components Components | Fascin | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / BETA-TREFOIL DOMAIN | ||||||
| Function / homology |  Function and homology information Function and homology informationmicrospike / parallel actin filament bundle assembly / regulation of microvillus assembly / positive regulation of extracellular matrix disassembly / establishment of apical/basal cell polarity / microspike assembly / cell projection membrane / podosome / positive regulation of podosome assembly / cell-cell junction assembly ...microspike / parallel actin filament bundle assembly / regulation of microvillus assembly / positive regulation of extracellular matrix disassembly / establishment of apical/basal cell polarity / microspike assembly / cell projection membrane / podosome / positive regulation of podosome assembly / cell-cell junction assembly / positive regulation of filopodium assembly / establishment or maintenance of cell polarity / microvillus / actin filament bundle assembly / positive regulation of lamellipodium assembly / stress fiber / ruffle / filopodium / cell motility / regulation of actin cytoskeleton organization / actin filament binding / cell-cell junction / cell migration / actin cytoskeleton / lamellipodium / protein-macromolecule adaptor activity / actin binding / cell cortex / growth cone / actin cytoskeleton organization / Interleukin-4 and Interleukin-13 signaling / cytoskeleton / cadherin binding / RNA binding / extracellular exosome / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2 Å | ||||||
 Authors Authors | Jansen, S. / Dominguez, R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2011 Title: Mechanism of actin filament bundling by fascin. Authors: Jansen, S. / Collins, A. / Yang, C. / Rebowski, G. / Svitkina, T. / Dominguez, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3p53.cif.gz | 401.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3p53.ent.gz | 330.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3p53.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p5/3p53ftp://data.pdbj.org/pub/pdb/validation_reports/p5/3p53 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | / Singed-like protein / 55 kDa actin-bundling protein / p55 Mass: 54868.152 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FAN1, FSCN1, HSN, Human, SNL / Plasmid: pTYB12 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q16658 Escherichia coli (E. coli) / References: UniProt: Q16658 |
|---|
-Non-polymers , 5 types, 411 molecules 


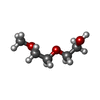

| #2: Chemical | ChemComp-GOL / Glycerol Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical | ChemComp-12P / | Polyethylene glycol Mass: 546.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H50O13 / Comment: precipitant*YM Mass: 546.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H50O13 / Comment: precipitant*YM#4: Chemical | ChemComp-1PE / Polyethylene glycol Mass: 238.278 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM Mass: 238.278 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM#5: Chemical | ChemComp-PG0 / | 2-(2-Methoxyethoxy)ethanol Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: inhibitor, precipitant*YM Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: inhibitor, precipitant*YM#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 396 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.29 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 24% PEG 3350, 0.2 M Lithium Acetate, 1mM DTT, 4% glycerol, pH 8, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 / Wavelength: 0.9795, 0.9798, 1.0 / Beamline: 17-ID / Wavelength: 1 / Wavelength: 0.9795, 0.9798, 1.0 | ||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 1, 2009 | ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 2→30.7 Å / Num. all: 65070 / Num. obs: 63704 / % possible obs: 97.9 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 6.9 % / Rmerge(I) obs: 0.057 / Net I/σ(I): 22.5 | ||||||||||||||||||
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 4.4 % / Rmerge(I) obs: 0.462 / Mean I/σ(I) obs: 2.6 / % possible all: 87.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2→30.661 Å / SU ML: 0.28 / σ(F): 1.44 / σ(I): 0 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.95 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 51.485 Å2 / ksol: 0.345 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→30.661 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
