 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3p06: Crystal structure of Tellina virus 1 VP4 protease in the form of ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3p06 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Tellina virus 1 VP4 protease in the form of an intra-molecular(cis)acyl-enzyme complex. | ||||||
 Components Components | VP4 protein | ||||||
 Keywords Keywords |  HYDROLASE / cis-cleavage / intramolecular acyl-enzyme / ester-linkage / alpha/beta protein / protease / polyprotein processing / acyl-enzyme HYDROLASE / cis-cleavage / intramolecular acyl-enzyme / ester-linkage / alpha/beta protein / protease / polyprotein processing / acyl-enzyme | ||||||
| Function / homology |  Function and homology information Function and homology informationserine-type peptidase activity / viral capsid / host cell cytoplasm / structural molecule activity / metal ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Tellina virus 1 Tellina virus 1 | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.1 Å | ||||||
 Authors Authors | Chung, I.Y.W. / Paetzel, M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2011 Title: Crystal Structure of a Viral Protease Intramolecular Acyl-enzyme Complex: INSIGHTS INTO cis-CLEAVAGE AT THE VP4/VP3 JUNCTION OF TELLINA BIRNAVIRUS. Authors: Chung, I.Y. / Paetzel, M. #1: Journal: Acta Crystallogr.,Sect.F / Year: 2011 Title: Expression, purification and crystallization of VP4 protease from Tellina virus 1. Authors: Chung, I.Y. / Paetzel, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3p06.cif.gz | 83.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3p06.ent.gz | 68.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3p06.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p0/3p06ftp://data.pdbj.org/pub/pdb/validation_reports/p0/3p06 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
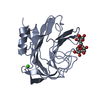
| Similar structure data |












-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 20563.883 Da / Num. of mol.: 1 / Fragment: UNP residues 637-830 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Tellina virus 1 / Gene: viral protein 4 (VP4) / Plasmid: pET28b+ / Production host:  Escherichia coli (E. coli) / Strain (production host): Tuner (DE3) / References: UniProt: Q2PBR5, EC: 3.4.21.115 Escherichia coli (E. coli) / Strain (production host): Tuner (DE3) / References: UniProt: Q2PBR5, EC: 3.4.21.115 |
|---|
-Non-polymers , 6 types, 54 molecules 


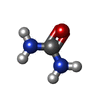


| #2: Chemical | ChemComp-SO4 / Sulfate Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical | Glycerol Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-BME / | 2-Mercaptoethanol Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS#5: Chemical | ChemComp-URE / | Urea Mass: 60.055 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH4N2O Mass: 60.055 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH4N2O#6: Chemical | ChemComp-CL / | Chloride Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 47 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 51.86 % |
|---|---|
| Crystal grow | Temperature: 296 K / pH: 5 Details: reservoir: 21% PEG8000, 0.55M ammonium sulfate. drop: On a coverslip, 1 microliter of VP4 was mixed with 1 microliter of reservoir reagent(21% PEG8000, 0.55M ammonium sulfate) and 1 ...Details: reservoir: 21% PEG8000, 0.55M ammonium sulfate. drop: On a coverslip, 1 microliter of VP4 was mixed with 1 microliter of reservoir reagent(21% PEG8000, 0.55M ammonium sulfate) and 1 microliter of 0.2M urea as additive. To aid in crystal nucleation, this drop was seeded with 1 microliter of selenomethionine- labelled crystal from an older drop, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 296K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.97893 / Beamline: 08ID-1 / Wavelength: 0.97893 |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: May 28, 2010 Details: DCM WITH CRYO-COOLED 1ST CRYSTAL SAGITTALLY BENT 2ND CRYSTAL FOLLOWED BY VERTICALLY FOCUSING MIRROR. |
| Radiation | Monochromator: A DOUBLE CRYSTAL MONOCHROMATOR (DCM) / Protocol: SAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97893 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→52 Å / Num. obs: 13466 / % possible obs: 99.8 % / Observed criterion σ(I): 2 / Redundancy: 11.4 % / Rmerge(I) obs: 0.107 / Net I/σ(I): 14.5 |
| Reflection shell | Resolution: 2.1→2.2 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 4.3 / Rsym value: 0.3 / % possible all: 98.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.1→52 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.928 / SU B: 9.221 / SU ML: 0.113 / Cross valid method: THROUGHOUT / ESU R Free: 0.171 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.44 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→52 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.15 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
