解像度: 3→28.75 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.854 / SU B: 37.362 / SU ML: 0.403 / 交差検証法: THROUGHOUT / ESU R Free: 0.496 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FILE CONTAINS FRIEDEL PAIRS.
Rfactor
反射数
%反射
Selection details
Rfree
0.28506
526
10 %
RANDOM
Rwork
0.20043
-
-
-
obs
0.20902
9113
100 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.2 Å / 溶媒モデル: MASK
原子変位パラメータ
Biso mean: 73.294 Å2
Baniso -1
Baniso -2
Baniso -3
1-
2.92 Å2
0 Å2
0 Å2
2-
-
2.92 Å2
0 Å2
3-
-
-
-5.83 Å2
精密化ステップ
サイクル: LAST / 解像度: 3→28.75 Å
タンパク質
核酸
リガンド
溶媒
全体
原子数
1377
0
21
84
1482
拘束条件
Refine-ID
タイプ
Dev ideal
Dev ideal target
数
X-RAY DIFFRACTION
r_bond_refined_d
0.05
0.022
1419
X-RAY DIFFRACTION
r_angle_refined_deg
3.918
1.997
1932
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
9.665
5
179
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
37.526
25.156
64
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
27.313
15
223
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
28.056
15
7
X-RAY DIFFRACTION
r_chiral_restr
0.207
0.2
218
X-RAY DIFFRACTION
r_gen_planes_refined
0.011
0.02
1098
X-RAY DIFFRACTION
r_nbd_refined
0.344
0.2
796
X-RAY DIFFRACTION
r_nbtor_refined
0.377
0.2
973
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.234
0.2
84
X-RAY DIFFRACTION
r_metal_ion_refined
0.243
0.2
7
X-RAY DIFFRACTION
r_symmetry_vdw_refined
0.384
0.2
31
X-RAY DIFFRACTION
r_symmetry_hbond_refined
0.277
0.2
1
X-RAY DIFFRACTION
r_symmetry_metal_ion_refined
0.007
0.2
1
X-RAY DIFFRACTION
r_mcbond_it
1.343
1.5
939
X-RAY DIFFRACTION
r_mcangle_it
2.234
2
1448
X-RAY DIFFRACTION
r_scbond_it
4.172
3
570
X-RAY DIFFRACTION
r_scangle_it
6.355
4.5
484
LS精密化 シェル
解像度: 3.002→3.079 Å / Total num. of bins used: 20
Rfactor
反射数
%反射
Rfree
0.389
37
-
Rwork
0.352
335
-
obs
-
-
100 %
精密化 TLS
手法: refined / Origin x: 0.9161 Å / Origin y: 16.0247 Å / Origin z: -23.4031 Å
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード peptide deformylase (ペプチドデホルミラーゼ) / 1B / PDF / N-terminal excision pathway / NME /
peptide deformylase (ペプチドデホルミラーゼ) / 1B / PDF / N-terminal excision pathway / NME /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード


















 PDBj
PDBj 集合体
集合体


 分子量: 65.409 Da / 分子数: 6 / 由来タイプ: 合成 / 式: Zn
分子量: 65.409 Da / 分子数: 6 / 由来タイプ: 合成 / 式: Zn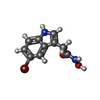
 分子量: 269.095 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H9BrN2O2
分子量: 269.095 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H9BrN2O2 分子量: 18.015 Da / 分子数: 84 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 84 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: PROXIMA 1 / 波長: 0.98 Å
/ ビームライン: PROXIMA 1 / 波長: 0.98 Å 解析
解析