 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3nop: Light-induced intermediate structure L1 of Pseudomonas aeruginosa... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3nop | ||||||
|---|---|---|---|---|---|---|---|
| Title | Light-induced intermediate structure L1 of Pseudomonas aeruginosa bacteriophytochrome | ||||||
 Components Components | Bacteriophytochrome | ||||||
 Keywords Keywords |  SIGNALING PROTEIN / intermediate structure / chromophore binding pocket / difference Fourier method SIGNALING PROTEIN / intermediate structure / chromophore binding pocket / difference Fourier method | ||||||
| Function / homology |  Function and homology information Function and homology informationosmosensory signaling via phosphorelay pathway / detection of visible light / phosphorelay response regulator activity / protein kinase activator activity / histidine kinase / photoreceptor activity / phosphorelay sensor kinase activity / regulation of DNA-templated transcription / ATP binding / identical protein bindingSimilarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / difference Fourier Method / Resolution: 2.8 Å | ||||||
 Authors Authors | Yang, X. / Ren, Z. / Moffat, K. | ||||||
 Citation Citation | Journal: Nature / Year: 2011 Title: Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Authors: Yang, X. / Ren, Z. / Kuk, J. / Moffat, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3nop.cif.gz | 108.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3nop.ent.gz | 83.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3nop.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/no/3nopftp://data.pdbj.org/pub/pdb/validation_reports/no/3nop | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3nhqSC  3notC  3nouC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 56823.230 Da / Num. of mol.: 1 Fragment: N-terminal photosensory core module, UNP residues 1-499 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Strain: PA01 / Gene: bphP, PA4117 / Plasmid: pET24 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 / References: UniProt: Q9HWR3, histidine kinase |
|---|---|
| #2: Chemical | ChemComp-BLA / 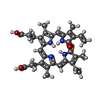  Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6 Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.03 Å3/Da / Density % sol: 59.4 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 7.7 Details: 10mg/ml protein, 0.1M Tris HCl buffer, 0.45M ammonium phosphate, pH 7.7, VAPOR DIFFUSION, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.97857 Å / Beamline: 21-ID-G / Wavelength: 0.97857 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Nov 12, 2008 |
| Radiation | Monochromator: C(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→50 Å / Num. all: 158473 / Num. obs: 131216 / % possible obs: 82.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5.5 % / Rmerge(I) obs: 0.118 |
- Processing
Processing
| Software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: difference Fourier Method Starting model: PDB ENTRY 3NHQ Resolution: 2.8→50 Å / Num. reflection all: 15473 / Num. reflection obs: 131216 / Occupancy max: 1 / Occupancy min: 0.56 / Cross valid method: real space correlation coefficient / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: This cryo-trapped structure was determined based on difference Fourier method. The L1 (3NOP), L2(3NOT) and L3(3NOU) structures were refined jointly in real space against a set of (Flight- ...Details: This cryo-trapped structure was determined based on difference Fourier method. The L1 (3NOP), L2(3NOT) and L3(3NOU) structures were refined jointly in real space against a set of (Flight-Fdark) difference maps representing mixtures of the L1, L2 and L3 structures in variable relative concentrations using software DynamiX. DynamiX is a collection of software tools for analyzing dynamic crystallographic data developed by Zhong Ren. Algorithms and methods are described in Ren, Z et al. Resolution of structural heterogeneity in dynamic and static crystallography. Manuscript in preparation. | ||||||||||||
| Displacement parameters | Biso max: 167.38 Å2 / Biso min: 22.56 Å2 | ||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→50 Å
|
