METAL BINDING PROTEIN / E3 ubiquitin ligase (ユビキチンリガーゼ) / UBR box / Zinc-binding protein (ジンクフィンガー) / N-end rule / Ligase (リガーゼ)
機能・相同性
機能・相同性情報
regulation of dipeptide transport / UBR1-RAD6 ubiquitin ligase complex / proteasome regulatory particle binding / stress-induced homeostatically regulated protein degradation pathway / ubiquitin-dependent protein catabolic process via the N-end rule pathway / cytoplasm protein quality control by the ubiquitin-proteasome system / mitochondria-associated ubiquitin-dependent protein catabolic process / Antigen processing: Ubiquitination & Proteasome degradation / proteasome regulatory particle, base subcomplex / ribosome-associated ubiquitin-dependent protein catabolic process ...regulation of dipeptide transport / UBR1-RAD6 ubiquitin ligase complex / proteasome regulatory particle binding / stress-induced homeostatically regulated protein degradation pathway / ubiquitin-dependent protein catabolic process via the N-end rule pathway / cytoplasm protein quality control by the ubiquitin-proteasome system / mitochondria-associated ubiquitin-dependent protein catabolic process / Antigen processing: Ubiquitination & Proteasome degradation / proteasome regulatory particle, base subcomplex / ribosome-associated ubiquitin-dependent protein catabolic process / ERAD pathway / protein monoubiquitination / ubiquitin ligase complex / RING-type E3 ubiquitin transferase / protein polyubiquitination / ubiquitin-protein transferase activity / ubiquitin protein ligase activity / protein ubiquitination / zinc ion binding / 細胞質 類似検索 - 分子機能
E3 ubiquitin-protein ligase UBR-like, C-terminal / Proteolysis_6 C-terminal / E3 ubiquitin-protein ligase UBR1-like / Putative zinc finger in N-recognin (UBR box) / Zinc finger, UBR-type / Zinc finger UBR-type profile. / Putative zinc finger in N-recognin, a recognition component of the N-end rule pathway 類似検索 - ドメイン・相同性
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード E3 ubiquitin ligase (ユビキチンリガーゼ) / UBR box /
E3 ubiquitin ligase (ユビキチンリガーゼ) / UBR box /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード













 PDBj
PDBj



 集合体
集合体


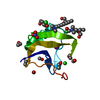
 分子量: 65.409 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Zn
分子量: 65.409 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Zn 分子量: 18.015 Da / 分子数: 22 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 22 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 4A / 波長: 1 Å
/ ビームライン: 4A / 波長: 1 Å 解析
解析