 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3llu: Crystal structure of the nucleotide-binding domain of Ras-related... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3llu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the nucleotide-binding domain of Ras-related GTP-binding protein C | ||||||
 Components Components | Ras-related GTP-binding protein C | ||||||
 Keywords Keywords |  HYDROLASE / Structural Genomics Consortium / SGC / Cytoplasm / GTP-binding / Nucleotide-binding / Nucleus / Phosphoprotein HYDROLASE / Structural Genomics Consortium / SGC / Cytoplasm / GTP-binding / Nucleotide-binding / Nucleus / Phosphoprotein | ||||||
| Function / homology |  Function and homology information Function and homology informationGtr1-Gtr2 GTPase complex / FNIP-folliculin RagC/D GAP / regulation of TORC1 signaling / protein localization to lysosome / regulation of TOR signaling / MTOR signalling / Amino acids regulate mTORC1 / Energy dependent regulation of mTOR by LKB1-AMPK / protein localization to membrane / enzyme-substrate adaptor activity ...Gtr1-Gtr2 GTPase complex / FNIP-folliculin RagC/D GAP / regulation of TORC1 signaling / protein localization to lysosome / regulation of TOR signaling / MTOR signalling / Amino acids regulate mTORC1 / Energy dependent regulation of mTOR by LKB1-AMPK / protein localization to membrane / enzyme-substrate adaptor activity / Macroautophagy / small GTPase-mediated signal transduction / mTORC1-mediated signalling / cellular response to nutrient levels / response to amino acid / protein-membrane adaptor activity / positive regulation of TORC1 signaling / cellular response to amino acid starvation / cellular response to starvation / negative regulation of autophagy / RNA splicing / Regulation of PTEN gene transcription / cellular response to amino acid stimulus / TP53 Regulates Metabolic Genes / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / protein localization / GDP binding / GTPase binding / lysosome / molecular adaptor activity / protein heterodimerization activity / lysosomal membrane / intracellular membrane-bounded organelle / GTPase activity / DNA-templated transcription / apoptotic process / GTP binding / magnesium ion binding / nucleoplasm / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.4 Å | ||||||
 Authors Authors | Nedyalkova, L. / Tempel, W. / Tong, Y. / Crombet, L. / Zhong, N. / Guan, X. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. ...Nedyalkova, L. / Tempel, W. / Tong, Y. / Crombet, L. / Zhong, N. / Guan, X. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. / Park, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: to be published Title: Crystal structure of the nucleotide-binding domain of Ras-related GTP-binding protein C Authors: Nedyalkova, L. / Tempel, W. / Tong, Y. / Crombet, L. / Zhong, N. / Guan, X. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. / Park, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3llu.cif.gz | 96.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3llu.ent.gz | 71.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3llu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ll/3lluftp://data.pdbj.org/pub/pdb/validation_reports/ll/3llu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2q3fS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22895.889 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: RRAGC / Plasmid: pET28-MHL / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21-V2R-pRARE2 / References: UniProt: Q9HB90 Escherichia coli (E. coli) / Strain (production host): BL21-V2R-pRARE2 / References: UniProt: Q9HB90 | ||
|---|---|---|---|
| #2: Chemical | ChemComp-GNP / 5'-Guanylyl imidodiphosphate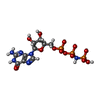  Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM | ||
| #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||
| #4: Chemical | ChemComp-UNX /   Num. of mol.: 9 / Source method: obtained synthetically Num. of mol.: 9 / Source method: obtained synthetically#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.16 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 2.0M ammonium sulfate, 0.2M sodium chloride, 0.1M HEPES. 1:100 (w/w) papain and GMPPNP were also added. Please note: mass spectral analysis of TEV protease digest prior to crystallization ...Details: 2.0M ammonium sulfate, 0.2M sodium chloride, 0.1M HEPES. 1:100 (w/w) papain and GMPPNP were also added. Please note: mass spectral analysis of TEV protease digest prior to crystallization suggested that removal of the His-tag was incomplete even after 2 days. However, without TEV protease treatment no crystals were obtained using otherwise identical crystallization conditions. Also note the presence of papain in the crystallization drop., pH 7.5, vapor diffusion, sitting drop, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97934 Å / Beamline: 19-ID / Wavelength: 0.97934 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC Q315 / Detector: CCD / Date: Dec 16, 2009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97934 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.4→25 Å / Num. obs: 36466 / % possible obs: 100 % / Redundancy: 7.3 % / Rmerge(I) obs: 0.055 / Χ2: 1.519 / Net I/σ(I): 12.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 2Q3F Resolution: 1.4→22.84 Å / Cor.coef. Fo:Fc: 0.969 / Cor.coef. Fo:Fc free: 0.963 / WRfactor Rfree: 0.182 / WRfactor Rwork: 0.155 / SU B: 1.644 / SU ML: 0.03 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.063 / ESU R Free: 0.056 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED. ARP/WARP, COOT and the MOLPROBITY server were also used during refinement.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.63 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→22.84 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 26.1668 Å / Origin y: 11.7144 Å / Origin z: 26.7548 Å
|
