 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3hpo: Crystal structure of fragment DNA polymerase I from Bacillus stea... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hpo | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of fragment DNA polymerase I from Bacillus stearothermophilus Y714S mutant bound to G:T mismatch | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSFERASE/DNA /  protein-DNA complex / DNA polymerase I / DNA replication / DNA-binding / DNA-directed DNA polymerase / Hydrolase / Nuclease / Nucleotidyltransferase / Transferase / TRANSFERASE-DNA COMPLEX protein-DNA complex / DNA polymerase I / DNA replication / DNA-binding / DNA-directed DNA polymerase / Hydrolase / Nuclease / Nucleotidyltransferase / Transferase / TRANSFERASE-DNA COMPLEX | |||||||||
| Function / homology |  Function and homology informationTaq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / 5' to 3' exonuclease, C-terminal subdomain / Ribonuclease H-like superfamily/Ribonuclease H / DNA polymerase; domain 1 / Nucleotidyltransferase; domain 5 / Alpha-Beta Plaits / Up-down Bundle / 2-Layer Sandwich ...Taq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / 5' to 3' exonuclease, C-terminal subdomain / Ribonuclease H-like superfamily/Ribonuclease H / DNA polymerase; domain 1 / Nucleotidyltransferase; domain 5 / Alpha-Beta Plaits / Up-down Bundle / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta Function and homology informationTaq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / 5' to 3' exonuclease, C-terminal subdomain / Ribonuclease H-like superfamily/Ribonuclease H / DNA polymerase; domain 1 / Nucleotidyltransferase; domain 5 / Alpha-Beta Plaits / Up-down Bundle / 2-Layer Sandwich ...Taq DNA Polymerase; Chain T, domain 4 / Taq DNA Polymerase; Chain T, domain 4 / Alpha-Beta Plaits - #370 / 5' to 3' exonuclease, C-terminal subdomain / Ribonuclease H-like superfamily/Ribonuclease H / DNA polymerase; domain 1 / Nucleotidyltransferase; domain 5 / Alpha-Beta Plaits / Up-down Bundle / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology | |||||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | |||||||||
 Authors Authors | Wu, E.Y. / Beese, L.S. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2011 Title: The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an "ajar" intermediate conformation in the nucleotide selection mechanism. Authors: Wu, E.Y. / Beese, L.S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hpo.cif.gz | 167.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hpo.ent.gz | 122.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3hpo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hp/3hpoftp://data.pdbj.org/pub/pdb/validation_reports/hp/3hpo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3hp6C  3ht3C  1l3uS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a complex of enzyme and DNA. The asymmetric unit is the same as the biological assembly. |
-Components
-DNA chain , 2 types, 2 molecules BC
| #2: DNA chain | Mass: 2684.789 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: Synthetic oligonucleotide primer strand |
|---|---|
| #3: DNA chain | Mass: 3101.028 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: Synthetic oligonucleotide template strand |
-Protein / Sugars , 2 types, 2 molecules A
| #1: Protein | Mass: 66128.859 Da / Num. of mol.: 1 / Fragment: Residues 297-876 / Mutation: D329A, Y714S Source method: isolated from a genetically manipulated source Details: Isolate from Idaho hot spring Source: (gene. exp.) Geobacillus stearothermophilus (bacteria)Gene: DPO1 / Plasmid: pET30a / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: DNA-directed DNA polymerase |
|---|---|
| #4: Polysaccharide | beta-D-fructofuranose-(2-1)-alpha-D-glucopyranose / sucrose / 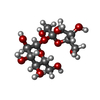  , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1 , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Details: oligosaccharide with reducing-end-to-reducing-end glycosidic bond References: sucrose |
-Non-polymers , 4 types, 727 molecules 



| #5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
|---|---|
| #6: Chemical | ChemComp-TTP / Thymidine triphosphate Mass: 482.168 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N2O14P3 Mass: 482.168 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N2O14P3 |
| #7: Chemical | ChemComp-SO4 / Sulfate Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #8: Water | ChemComp-HOH / WaterMass: 18.015 Da / Num. of mol.: 724 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.1 Å3/Da / Density % sol: 60.34 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: 50% Saturated ammonium sulfate, 0.1M MES, 2.5% v/v Methylpentanediol, 10mM Magnesium sulfate, pH 5.8, VAPOR DIFFUSION, HANGING DROP, temperature 290K | ||||||||||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-BM / Wavelength: 1 Å / Beamline: 22-BM / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Aug 1, 2008 |
| Radiation | Monochromator: Si(111) crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.74→45.98 Å / Num. all: 92241 / Num. obs: 83554 / % possible obs: 90.6 % / Observed criterion σ(I): -3 / Redundancy: 5 % / Biso Wilson estimate: 29.055 Å2 / Rmerge(I) obs: 0.084 / Net I/σ(I): 10.66 |
| Reflection shell | Resolution: 1.74→1.85 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.409 / Mean I/σ(I) obs: 2.9 / Num. measured obs: 53772 / Num. unique all: 11929 / Num. unique obs: 11929 / % possible all: 77.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1L3U Resolution: 1.75→45.98 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.923 / WRfactor Rfree: 0.251 / WRfactor Rwork: 0.207 / Occupancy max: 1 / Occupancy min: 0.3 / FOM work R set: 0.845 / SU B: 2.4 / SU ML: 0.077 / SU R Cruickshank DPI: 0.116 / SU Rfree: 0.116 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.116 / ESU R Free: 0.116 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. U VALUES: REFINED INDIVIDUALLY.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 73.36 Å2 / Biso mean: 25.3 Å2 / Biso min: 6.78 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→45.98 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.795 Å / Total num. of bins used: 20
|
