 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3emy: Crystal structure of Trichoderma reesei aspartic proteinase compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3emy | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Trichoderma reesei aspartic proteinase complexed with pepstatin A | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR /  Trichoderma reesei / aspartic proteinase / Aspartyl protease / Protease / HYDROLASE-HYDROLASE INHIBITOR complex Trichoderma reesei / aspartic proteinase / Aspartyl protease / Protease / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||||||||
| Function / homology |  Function and homology information Function and homology information | ||||||||||||
| Biological species |  Hypocrea jecorina (fungus) Hypocrea jecorina (fungus) Streptomyces argenteolus subsp. toyonakensis (bacteria) Streptomyces argenteolus subsp. toyonakensis (bacteria) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||||||||
 Authors Authors | Nascimento, A.S. / Krauchenco, S. / Golubev, A.M. / Gustchina, A. / Wlodawer, A. / Polikarpov, I. | ||||||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2008 Title: Statistical coupling analysis of aspartic proteinases based on crystal structures of the Trichoderma reesei enzyme and its complex with pepstatin A. Authors: Nascimento, A.S. / Krauchenco, S. / Golubev, A.M. / Gustchina, A. / Wlodawer, A. / Polikarpov, I. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3emy.cif.gz | 159.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3emy.ent.gz | 132.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3emy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/em/3emyftp://data.pdbj.org/pub/pdb/validation_reports/em/3emy | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34399.152 Da / Num. of mol.: 1 / Fragment: UNP residues 79-407 / Source method: isolated from a natural source / Source: (natural) Hypocrea jecorina (fungus) / References: UniProt: Q2WBH2 |
|---|---|
| #2: Protein/peptide | / 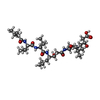  Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 685.891 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: This sequence occurs naturally in Streptomyces Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 685.891 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: This sequence occurs naturally in StreptomycesSource: (synth.) Streptomyces argenteolus subsp. toyonakensis (bacteria)References: Pepstatin |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 618 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 618 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 60.9 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 15% PEG3350, 50MM potassium phosphate buffer, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LNLS  / Beamline: D03B-MX1 / Wavelength: 1.5 Å / Beamline: D03B-MX1 / Wavelength: 1.5 Å |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Jan 1, 2006 |
| Radiation | Monochromator: SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→19.468 Å / Num. obs: 37688 / % possible obs: 96.3 % / Observed criterion σ(F): 1.38 / Observed criterion σ(I): 1.38 |
| Reflection shell | Resolution: 1.85→1.89 Å / % possible all: 95 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.85→19.468 Å / SU ML: 0.21 / σ(F): 1.35 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 70.835 Å2 / ksol: 0.363 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→19.468 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 8.4035 Å / Origin y: 54.3117 Å / Origin z: 7.5643 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
