 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3cz7: Molecular Basis for the Autoregulation of the Protein Acetyl Tran... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3cz7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Molecular Basis for the Autoregulation of the Protein Acetyl Transferase Rtt109 | ||||||
 Components Components | Regulator of Ty1 transposition protein 109 | ||||||
 Keywords Keywords |  REPLICATION / Chromatin Stability REPLICATION / Chromatin Stability | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H3K23 acetyltransferase activity / histone H3K56 acetyltransferase activity / H3 histone acetyltransferase complex / DNA replication-dependent chromatin disassembly / histone H3K14 acetyltransferase activity / regulation of double-strand break repair via nonhomologous end joining / histone H3K9 acetyltransferase activity / maintenance of rDNA / replication-born double-strand break repair via sister chromatid exchange / histone H3 acetyltransferase activity ...histone H3K23 acetyltransferase activity / histone H3K56 acetyltransferase activity / H3 histone acetyltransferase complex / DNA replication-dependent chromatin disassembly / histone H3K14 acetyltransferase activity / regulation of double-strand break repair via nonhomologous end joining / histone H3K9 acetyltransferase activity / maintenance of rDNA / replication-born double-strand break repair via sister chromatid exchange / histone H3 acetyltransferase activity / retrotransposon silencing / histone H3K27 acetyltransferase activity / peptide-lysine-N-acetyltransferase activity / histone acetyltransferase / protein modification process / nucleosome assembly / regulation of gene expression / DNA damage response / chromatin / regulation of transcription by RNA polymerase II / nucleusSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2 Å | ||||||
 Authors Authors | Hoelz, A. / Stavropoulos, P. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2008 Title: Molecular basis for the autoregulation of the protein acetyl transferase Rtt109 Authors: Stavropoulos, P. / Nagy, V. / Blobel, G. / Hoelz, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3cz7.cif.gz | 89.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3cz7.ent.gz | 72.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3cz7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cz/3cz7ftp://data.pdbj.org/pub/pdb/validation_reports/cz/3cz7 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42181.465 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Gene: RTT109, KIM2, REM50 / Cell line (production host): BL21-CodonPlus(DE3)-RIL / Production host:  Escherichia coli (E. coli) / References: UniProt: Q07794 Escherichia coli (E. coli) / References: UniProt: Q07794 |
|---|---|
| #2: Chemical | ChemComp-ACO / Acetyl-CoA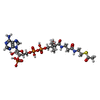  Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 276 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 276 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.72 % Description: THE STRUCTURE FACTOR FILE CONTAINS FRIEDEL PAIRS. |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 4.4 Details: 12% (w/v) PEG 4,000, 1 mM ZnCl2, and 100 mM Sodium Acetate, pH 4.4, temperature 294K, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X3A / Wavelength: 0.9798 / Beamline: X3A / Wavelength: 0.9798 |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Sep 20, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9798 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. obs: 55305 / Observed criterion σ(I): 3 |
- Processing
Processing
| Software | Name: REFMAC / Version: 5.2.0005 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2→30 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.951 / SU B: 4.85 / SU ML: 0.083 / TLS residual ADP flag: LIKELY RESIDUAL / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FRIEDEL PAIRS WERE USED FOR PHASING.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.46 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.05 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
