 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bre | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of P.aeruginosa PA3702 | ||||||
 Components Components | Probable two-component response regulator | ||||||
 Keywords Keywords |  SIGNALING PROTEIN / protein-nucleotide complex SIGNALING PROTEIN / protein-nucleotide complex | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of bacterial-type flagellum-dependent cell motility / diguanylate cyclase / diguanylate cyclase activity / cell adhesion involved in single-species biofilm formation / phosphorelay signal transduction system / nucleotide binding / identical protein binding / metal ion binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.4 Å | ||||||
 Authors Authors | De, N. / Pirruccello, M. / Krasteva, P.V. / Bae, N. / Raghavan, R.V. / Sondermann, H. | ||||||
 Citation Citation | Journal: Plos Biol. / Year: 2008 Title: Phosphorylation-independent regulation of the diguanylate cyclase WspR. Authors: De, N. / Pirruccello, M. / Krasteva, P.V. / Bae, N. / Raghavan, R.V. / Sondermann, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bre.cif.gz | 142 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bre.ent.gz | 110.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3bre.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/br/3breftp://data.pdbj.org/pub/pdb/validation_reports/br/3bre | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1w25S S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | two C2-crystal symmetry-related dimers (chains A,B) form a biological unit (tetramer) |
-Components
| #1: Protein | Mass: 39213.250 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Strain: PAO1 / Gene: wspR / Plasmid: pET21a / Species (production host): Escherichia coli / Production host: Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: Q9HXT9#2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-C2E / Cyclic di-GMP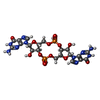  Mass: 690.411 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H24N10O14P2 Mass: 690.411 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H24N10O14P2 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.32 Å3/Da / Density % sol: 63 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 0.1 M Tris-Cl pH8.0, 2.9M NaCl, 15% xylitol, VAPOR DIFFUSION, HANGING DROP, temperature 294K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.9771 Å / Beamline: A1 / Wavelength: 0.9771 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Nov 18, 2006 / Details: mirrors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Horizontal focusing 5.05 asymmetric cut Si(111) Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9771 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 4.3 % / Av σ(I) over netI: 12.3 / Number: 171005 / Rmerge(I) obs: 0.066 / Χ2: 1.01 / D res high: 2.39 Å / D res low: 50 Å / Num. obs: 40110 / % possible obs: 98.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.39→50 Å / Num. obs: 40110 / % possible obs: 98.9 % / Redundancy: 4.3 % / Biso Wilson estimate: 45.2 Å2 / Rmerge(I) obs: 0.066 / Rsym value: 0.049 / Χ2: 1.012 / Net I/σ(I): 12.3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.39→2.48 Å / Redundancy: 3 % / Rmerge(I) obs: 0.459 / Mean I/σ(I) obs: 2 / Num. unique all: 3803 / Rsym value: 0.459 / Χ2: 0.871 / % possible all: 94.5 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1W25 Resolution: 2.4→33.78 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 84696.5 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 45.73 Å2 / ksol: 0.35 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 67.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→33.78 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
