 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 3anp | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Crystal structure of Thermus thermophilus FadR, a TetR familly transcriptional repressor, in complex with lauroyl-CoA. | ||||||
 要素 要素 | Transcriptional repressor, TetR family | ||||||
 キーワード キーワード |  TRANSCRIPTION (転写 (生物学)) / All Alpha Protein / Transcriptional Repressor / DNA (デオキシリボ核酸) / Acyl-CoA (アシルCoA) TRANSCRIPTION (転写 (生物学)) / All Alpha Protein / Transcriptional Repressor / DNA (デオキシリボ核酸) / Acyl-CoA (アシルCoA) | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |   Thermus thermophilus (サーマス・サーモフィルス) Thermus thermophilus (サーマス・サーモフィルス) | ||||||
| 手法 | X線回折 / シンクロトロン / 単波長異常分散 / 解像度: 1.95 Å | ||||||
 データ登録者 データ登録者 | Agari, Y. / Agari, K. / Sakamoto, K. / Kuramitsu, S. / Shinkai, A. | ||||||
 引用 引用 | ジャーナル: Microbiology / 年: 2011 タイトル: TetR-family transcriptional repressor Thermus thermophilus FadR controls fatty acid degradation. 著者: Agari, Y. / Agari, K. / Sakamoto, K. / Kuramitsu, S. / Shinkai, A. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 3anp.cif.gz | 184.6 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb3anp.ent.gz | 154.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 3anp.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/an/3anpftp://data.pdbj.org/pub/pdb/validation_reports/an/3anp | HTTPS FTP |
|---|
-関連構造データ











-リンク
 PDBj
PDBj


- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 23711.828 Da / 分子数: 4 / 断片: UNP Residues 2-203 / 由来タイプ: 組換発現 由来: (組換発現) Thermus thermophilus (サーマス・サーモフィルス)株: HB8 / 遺伝子: FadR / プラスミド: pET-11a / 発現宿主: Escherichia coli (大腸菌) / 株 (発現宿主): B834(DE3) / 参照: UniProt: Q5SM42#2: 化合物 |   分子量: 949.837 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C33H58N7O17P3S 分子量: 949.837 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C33H58N7O17P3S#3: 化合物 | ChemComp-DAO / | ラウリン酸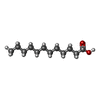  分子量: 200.318 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H24O2 分子量: 200.318 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H24O2#4: 水 | ChemComp-HOH / | 水 分子量: 18.015 Da / 分子数: 713 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 713 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 2 |
|---|
- 試料調製
試料調製
| 結晶 |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 |
|
-データ収集
| 回折 | 平均測定温度: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: SPring-8  / ビームライン: BL26B2 / 波長: 1 Å / ビームライン: BL26B2 / 波長: 1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 検出器 | タイプ: MARMOSAIC 225 mm CCD / 検出器: CCD / 日付: 2009年2月23日 詳細: vertically bent two dimensional focusing mirror coated in Rhodium | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 放射 | モノクロメーター: Fixed exit Si double crystal monochromator プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 放射波長 | 波長: 1 Å / 相対比: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | 冗長度: 6.6 % / Av σ(I) over netI: 33.4 / 数: 347357 / Rmerge(I) obs: 0.09 / Χ2: 1.41 / D res high: 2.25 Å / D res low: 50 Å / Num. obs: 53003 / % possible obs: 98.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 反射 | 解像度: 1.95→50 Å / Num. all: 81796 / Num. obs: 81796 / % possible obs: 99.1 % / 冗長度: 7.2 % / Biso Wilson estimate: 21.8 Å2 / Rmerge(I) obs: 0.075 / Χ2: 1.302 / Net I/σ(I): 11.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 反射 シェル | Diffraction-ID: 1
|
-位相決定
| 位相決定 | 手法: 単波長異常分散 | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing dm | FOM : 0.65 / FOM acentric: 0.65 / FOM centric: 0.68 / 反射: 48664 / Reflection acentric: 44564 / Reflection centric: 4100 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 単波長異常分散 / 解像度: 1.95→26.09 Å / Rfactor Rfree error: 0.003 / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.8365 / Data cutoff high absF: 2615321 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber / 詳細: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 53.9792 Å2 / ksol: 0.37 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso max: 90.4 Å2 / Biso mean: 25.0904 Å2 / Biso min: 8.02 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.95→26.09 Å
| ||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.95→2.07 Å / Rfactor Rfree error: 0.008 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
