 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3a6v | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the MutT protein in MN(II) bound holo form | ||||||
 Components Components | Mutator mutT protein | ||||||
 Keywords Keywords |  HYDROLASE / ALPHA-BETA-ALPHA SANDWICH / DNA DAMAGE / DNA REPAIR / DNA REPLICATION / MUTATOR PROTEIN HYDROLASE / ALPHA-BETA-ALPHA SANDWICH / DNA DAMAGE / DNA REPAIR / DNA REPLICATION / MUTATOR PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationdGDP catabolic process / 8-oxo-dGTP diphosphatase / 8-oxo-GDP phosphatase activity / 8-oxo-dGDP phosphatase activity / dGTP catabolic process / 8-oxo-7,8-dihydrodeoxyguanosine triphosphate pyrophosphatase activity / 8-oxo-7,8-dihydroguanosine triphosphate pyrophosphatase activity / nucleotide-excision repair / manganese ion binding / DNA replication ...dGDP catabolic process / 8-oxo-dGTP diphosphatase / 8-oxo-GDP phosphatase activity / 8-oxo-dGDP phosphatase activity / dGTP catabolic process / 8-oxo-7,8-dihydrodeoxyguanosine triphosphate pyrophosphatase activity / 8-oxo-7,8-dihydroguanosine triphosphate pyrophosphatase activity / nucleotide-excision repair / manganese ion binding / DNA replication / DNA repair / magnesium ion bindingSimilarity search - Function | ||||||
| Biological species |  Escherichia coli K-12 (bacteria) Escherichia coli K-12 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Nakamura, T. / Yamagata, Y. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2010 Title: Structural and dynamic features of the MutT protein in the recognition of nucleotides with the mutagenic 8-oxoguanine base Authors: Nakamura, T. / Meshitsuka, S. / Kitagawa, S. / Abe, N. / Yamada, J. / Ishino, T. / Nakano, H. / Tsuzuki, T. / Doi, T. / Kobayashi, Y. / Fujii, S. / Sekiguchi, M. / Yamagata, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3a6v.cif.gz | 66.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3a6v.ent.gz | 48.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3a6v.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a6/3a6vftp://data.pdbj.org/pub/pdb/validation_reports/a6/3a6v | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3a6sSC  3a6tC  3a6uC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14945.029 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli K-12 (bacteria) / Strain: K12 / Gene: MUTT / Plasmid: PET8C / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3)References: UniProt: P08337, Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides#2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#3: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#4: Chemical | Tartaric acid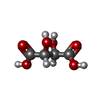  Mass: 150.087 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H6O6#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 131 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 131 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 50.09 % |
|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 1.4M POTASSIUM SODIUM TARTRATE, 87mM HEPES, pH 7.50, VAPOR DIFFUSION, HANGING DROP, temperature 288K |
-Data collection
| Diffraction | Mean temperature: 289 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: BL-18B / Wavelength: 1 Å / Beamline: BL-18B / Wavelength: 1 Å |
| Detector | Type: WEISSENBERG / Detector: DIFFRACTOMETER / Date: Dec 1, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→40 Å / Num. obs: 19810 / % possible obs: 98.1 % / Redundancy: 3.4 % / Biso Wilson estimate: 6 Å2 / Rmerge(I) obs: 0.042 / Net I/σ(I): 28.6 |
| Reflection shell | Resolution: 2→2.03 Å / Rmerge(I) obs: 0.064 / Mean I/σ(I) obs: 18.6 / % possible all: 96.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3A6S Resolution: 2→19.94 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1116098.45 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 41.81 Å2 / ksol: 0.33 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.94 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.013 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
