 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 376d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | A ZIPPER-LIKE DNA DUPLEX D(GCGAAAGCT) | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords Keywords DNA / RIGHT HANDED DNA / DOUBLE HELIX / OVERHANGING BASE / MODIFIED / MISMATCHED DNA / RIGHT HANDED DNA / DOUBLE HELIX / OVERHANGING BASE / MODIFIED / MISMATCHEDFunction / homology | COBALT HEXAMMINE(III) / | DNA Function and homology information Function and homology informationMethod | X-RAY DIFFRACTION / SYNCHROTRON / MAD AT BROMINE EDGE / Resolution: 2.1 Å  Authors AuthorsCruse, W.B.T. / Shepard, W. / Prange, T. / delalFortelle, E. / Fourme, R. |  CitationJournal: Structure / Year: 1998 CitationJournal: Structure / Year: 1998Title: A zipper-like duplex in DNA: the crystal structure of d(GCGAAAGCT) at 2.1 A resolution. Authors: Shepard, W. / Cruse, W.B. / Fourme, R. / de la Fortelle, E. / Prange, T. #1: Journal: Science / Year: 1997Title: RNA Tertiary Structure Mediation by Adenosine Platforms Authors: Cate, J.H. / Gooding, A.R. / Podell, E. / Zhou, K. / Golden, B.L. / Szewczak, A.A. / Kundrot, C.E. / Cech, T.R. / Doudna, J.A. #2: Journal: Structure / Year: 1997Title: Solution Structure of a Metal-binding Site in the Major Groove of RNA Complexed with Cobalt(III) Hexammine Authors: Kieft, J.S. / Tinoco Jr., I. #3: Journal: Nucleic Acids Res. / Year: 1989Title: Extraordinary Stable Structure of Short Single-Stranded DNA Fragments Containing a Specific Base Sequence: d(GCGAAAGC) Authors: Hirao, I. / Nichimura, Y. / Naraoka, T. / Watanabe, K. / Arata, Y. / Miura, K. #4: Journal: Biochemistry / Year: 1985Title: Structural Basis for Stabilisation of Z-DNA by Cobalt Haxaammine and Magnesium Cations Authors: Geissner, R.V. / Quigley, G.J. / Wang, H.J. / van der Marel, A. / van Boom, J.H. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 376d.cif.gz | 15.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb376d.ent.gz | 9.3 KB | Display | PDB format |
| PDBx/mmJSON format | 376d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/76/376dftp://data.pdbj.org/pub/pdb/validation_reports/76/376d | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 2843.733 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source |
|---|---|
| #2: Chemical | ChemComp-NCO / 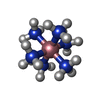  Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6 |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 36 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Components of the solutions |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 36 % | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 276 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW21B / Beamline: DW21B |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1996 / Details: DOUBLE MONO + MULTILAYER FOCUS |
| Radiation | Monochromator: SI(III) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.1→15 Å / Num. obs: 1832 / % possible obs: 95 % / Observed criterion σ(I): 3 / Rmerge(I) obs: 0.053 / Net I/σ(I): 11.3 |
| Reflection shell | Highest resolution: 2.1 Å / Rmerge(I) obs: 0.148 / Mean I/σ(I) obs: 4.2 / % possible all: 61 |
| Reflection | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 15 Å / % possible obs: 95 % / Observed criterion σ(I): 3 / Redundancy: 11.6 % / Num. measured all: 21345 |
| Reflection shell | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 2.2 Å / Mean I/σ(I) obs: 4.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD AT BROMINE EDGE Starting model: MANUALLY BUILT IN THE EXPERIMENTAL DENSITY Resolution: 2.1→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.15 Å / Luzzati d res low obs: 15 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 15 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d |
