Entry Database : PDB / ID : 2y2fTitle Crystal structure of Yersinia pestis YopH in complex with an aminooxy- containing platform compound for inhibitor design PROTEIN-TYROSINE PHOSPHATASE YOPH Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species YERSINIA PESTIS (bacteria)Method / / / Resolution : 1.78 Å Authors Lountos, G.T. / Bahta, M. / Dyas, B. / Ulrich, R.G. / Waugh, D.S. / Burke, T.R. Journal : J.Med.Chem. / Year : 2011Title : Utilization of Nitrophenylphosphates and Oxime-Based Ligation for the Development of Nanomolar Affinity Inhibitors of the Yersinia Pestis Outer Protein H (Yoph) Phosphatase.Authors : Bahta, M. / Lountos, G.T. / Dyas, B. / Kim, S. / Ulrich, R.G. / Waugh, D.S. / Burke, T.R. History Deposition Dec 14, 2010 Deposition site / Processing site Revision 1.0 Mar 16, 2011 Provider / Type Revision 1.1 Sep 28, 2011 Group / Version format complianceRevision 1.2 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords HYDROLASE /
HYDROLASE /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly
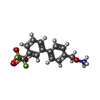
 Mass: 330.244 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H15F2NO4P
Mass: 330.244 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H15F2NO4P Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 22-ID / Wavelength: 1
/ Beamline: 22-ID / Wavelength: 1  Processing
Processing