 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2xo8: Crystal Structure of Myosin-2 in Complex with Tribromodichloropse... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xo8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Myosin-2 in Complex with Tribromodichloropseudilin | ||||||
 Components Components | MYOSIN-2 HEAVY CHAIN | ||||||
 Keywords Keywords | MOTOR PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationcalcium-dependent ATPase activity / pseudopodium retraction / uropod retraction / cytoplasmic actin-based contraction involved in forward cell motility / phagocytic cup base / pathogen-containing vacuole / response to differentiation-inducing factor 1 / equatorial cell cortex / RHO GTPases activate PAKs / contractile actin filament bundle assembly ...calcium-dependent ATPase activity / pseudopodium retraction / uropod retraction / cytoplasmic actin-based contraction involved in forward cell motility / phagocytic cup base / pathogen-containing vacuole / response to differentiation-inducing factor 1 / equatorial cell cortex / RHO GTPases activate PAKs / contractile actin filament bundle assembly / cell trailing edge / contractile vacuole organization / myosin filament assembly / aggregation involved in sorocarp development / culmination involved in sorocarp development / adenyl nucleotide binding / actomyosin contractile ring / hypotonic response / uropod / actin-myosin filament sliding / detection of mechanical stimulus / apical cortex / negative regulation of actin filament polymerization / bleb assembly / actomyosin / myosin filament / filopodium assembly / myosin II complex / early phagosome / microfilament motor activity / cortical actin cytoskeleton organization / cortical actin cytoskeleton / pseudopodium / cleavage furrow / cytoskeletal motor activity / mitotic cytokinesis / response to mechanical stimulus / response to cAMP / extracellular matrix / 14-3-3 protein binding / cell motility / response to hydrogen peroxide / protein localization / chemotaxis / actin filament binding / cell cortex / regulation of cell shape / cytoplasmic vesicle / cytoskeleton / calmodulin binding / ATP binding / identical protein binding / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  DICTYOSTELIUM DISCOIDEUM (eukaryote) DICTYOSTELIUM DISCOIDEUM (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Preller, M. / Chinthalapudi, K. / Martin, R. / Knoelker, H.J. / Manstein, D.J. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2011 Title: Inhibition of Myosin ATPase Activity by Halogenated Pseudilins: A Structure-Activity Study. Authors: Preller, M. / Chinthalapudi, K. / Martin, R. / Knolker, H. / Manstein, D.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xo8.cif.gz | 182.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xo8.ent.gz | 142.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2xo8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xo/2xo8ftp://data.pdbj.org/pub/pdb/validation_reports/xo/2xo8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2jj9S S: Starting model for refinement |
|---|---|
| Similar structure data |







































-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / MYOSIN II HEAVY CHAIN Mass: 88454.891 Da / Num. of mol.: 1 / Fragment: MOTOR DOMAIN, RESIDUES 3-761 Source method: isolated from a genetically manipulated source Source: (gene. exp.) DICTYOSTELIUM DISCOIDEUM (eukaryote) / Production host: DICTYOSTELIUM DISCOIDEUM (eukaryote) / References: UniProt: P08799 |
|---|---|
| #2: Chemical | ChemComp-H70 /   Mass: 464.763 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H4Br3Cl2NO Mass: 464.763 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H4Br3Cl2NO |
| #3: Chemical | ChemComp-AD9 / 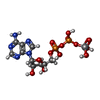  Mass: 527.149 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P2V Mass: 527.149 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P2V |
| #4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 435 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 435 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.8 Details: 50 MM HEPES(PH 6.8), 110 MM NACL, 11% PEG 5K-MME, 2% MPD, 5MM DTT, 1MM EGTA,5MM MGCL2 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X13 / Wavelength: 0.8055 / Beamline: X13 / Wavelength: 0.8055 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Nov 5, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8055 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→19.7 Å / Num. obs: 39289 / % possible obs: 100 % / Observed criterion σ(I): 2 / Redundancy: 8.2 % / Biso Wilson estimate: 31.8 Å2 / Rmerge(I) obs: 0.14 |
| Reflection shell | Resolution: 2.4→2.5 Å / Redundancy: 8.4 % / Rmerge(I) obs: 0.55 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2JJ9 Resolution: 2.4→19.7 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 5418533.32 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 57.1964 Å2 / ksol: 0.4 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 52 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→19.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.013 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
